今天小果带来的分享非常实用,在泛癌纯生信文章中必备的分析内容之一,用分裂小提琴图展示某个基因在TCGA肿瘤和正常组织中的表达差异,如果想要如此高颜值的图片出现在你的文章中,马上行动起来,跟着小果开始今天的实操学习吧!
- 数据下载
在进行数据分析之前,小果先带着大家进行泛癌表达矩阵和样本信息相关文件下载,通过该网址https://xenabrowser.net/datapages/下载,网页如下图:

2.准备需要的R包
#安装需要的R包
install.packages(“stringr”)
install.packages(“dplyr”)
install.packages(“ggplot2”)
install.packages(“RColorBrewer”)
#加载需要的R包
library(stringr)
library(dplyr)
library(ggplot2)
library(RColorBrewer)
targetGene <- “TP53” # 想要画的目标基因
3.输入文件读取和处理
#samplepair.txt,TCGA跟GTEx的组织对应关系文件
samplepair <- read.delim(“samplepair.txt”,as.is = T)
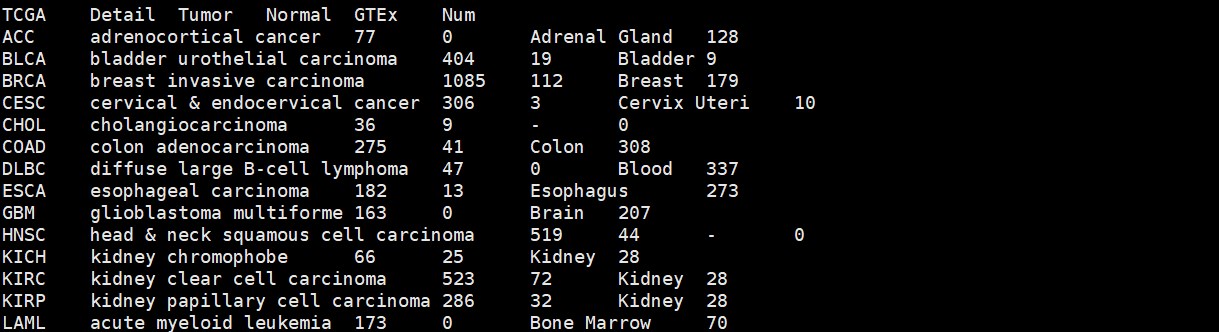
gtexpair <- samplepair[,c(1,2,5)]
gtexpair
#以TCGA的缩写为GTEx的组织命名
gtexpair$type <- paste0(gtexpair$TCGA,”_normal_GTEx”)
gtexpair$type2 <-“normal”
gtextcga <- gtexpair[,c(1,3:5)] #筛掉Detail列
colnames(gtextcga)[1:2] <- c(“tissue”,”X_primary_site”)
head(gtextcga)
#为GTEx的sample标出组织
#GTEX_phenotype.gz,GTEX样本和组织对应关系文件
gtexcase <- read.delim(file=”GTEX_phenotype.gz”,header=T,as.is = T)
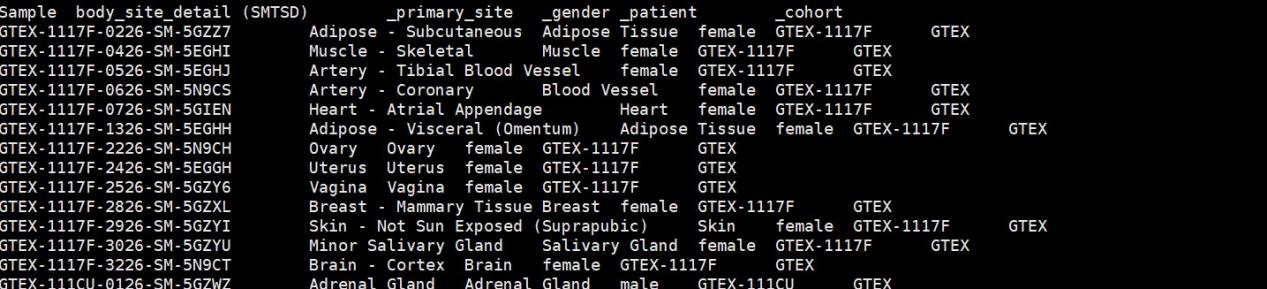
colnames(gtexcase)[1] <- “sample”
gtexcase2tcga <- merge(gtextcga,gtexcase,by=”X_primary_site”)
gtextable <- gtexcase2tcga[,c(5,2:4)]
head(gtextable)
tissue <- gtexpair$TCGA
names(tissue) <- gtexpair$Detail
#TCGA_phenotype_denseDataOnlyDownload.tsv.gz,TCGA样本与组织对应关系
tcgacase <- read.delim(file=”TCGA_phenotype_denseDataOnlyDownload.tsv.gz”,header=T,as.is = T)
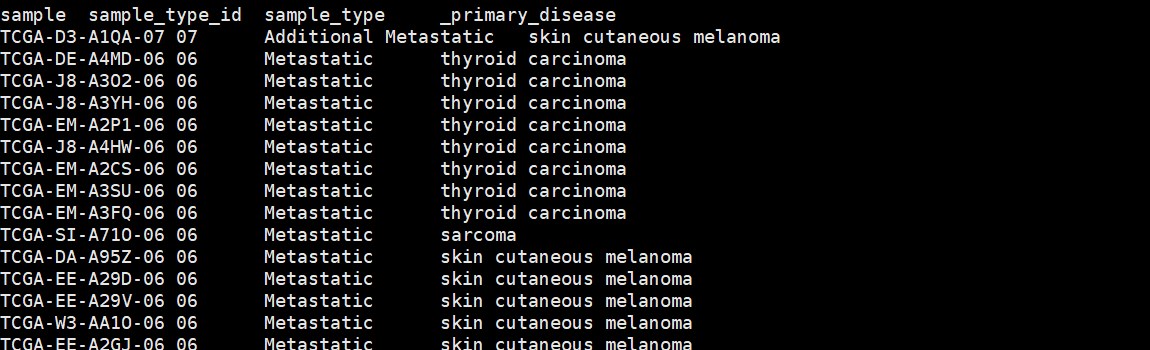
tcgacase$tissue <- tissue[tcgacase$X_primary_disease]
tcgacase$type<-ifelse(tcgacase$sample_type==’Solid Tissue Normal’,paste(tcgacase$tissue,”normal_TCGA”,sep=”_”),paste(tcgacase$tissue,”tumor_TCGA”,sep=”_”))
tcgacase$type2 <- ifelse(tcgacase$sample_type==’Solid Tissue Normal’,”normal”,”tumor”)
tcgatable<-tcgacase[,c(1,5:7)]
head(tcgatable)
#gtex泛癌基因表达矩阵文件,第一列为基因名,其他列为样本名
headgtex <- read_part(“gtex_RSEM_gene_tpm.gz”, rows = 1)
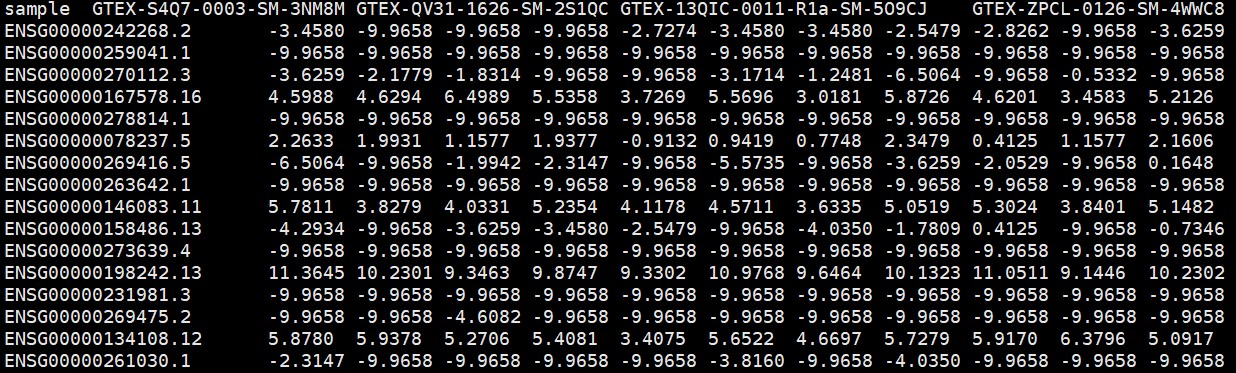
gtexsample <- headgtex[1,]
gtextable2 <- gtextable[gtextable$sample %in% gtexsample,]
#TCGA泛癌基因表达矩阵文件,第一列为基因名,其他列为样本名
headtcga <- read_part(“tcga_RSEM_gene_tpm.gz”, rows = 1)
tcgasample <- headtcga[1,]
tcgatable2 <- tcgatable[tcgatable$sample %in% tcgasample,]
tcga_gtex<-rbind(tcgatable2,gtextable2)
statable<-data.frame()
for (tissuer in sort(unique(tcga_gtex$tissue))){
tcga_gtex_tissuer<-tcga_gtex[tcga_gtex$tissue==tissuer,]
tissuer_tumor_TCGA <- paste0(tissuer,”_tumor_TCGA”)
tissuer_normal_TCGA <- paste0(tissuer,”_normal_TCGA”)
tissuer_normal_GTEx <- paste0(tissuer,”_normal_GTEx”)
tissuerdf<-data.frame(TCGA=tissuer,
TCGA_new_tumor=sum(tcga_gtex_tissuer$type==tissuer_tumor_TCGA),
TCGA_new_normal=sum(tcga_gtex_tissuer$type==tissuer_normal_TCGA),
GTEx_new_num=sum(tcga_gtex_tissuer$type==tissuer_normal_GTEx),
stringsAsFactors=F)
#str(tissuerdf)
statable<-rbind(statable,tissuerdf)
}
cbind(statable, samplepair[,c(1,3:6)])
idmap <- read.delim(“gencode.v23.annotation.gene.probemap”,as.is=T)
head(idmap)
# 读取第一列, 所有行
tpmid2row <- read_part(“gtex_RSEM_gene_tpm.gz”, rows = -1, columns = 1)
tpmid2row <- tpmid2row[2:nrow(tpmid2row)]
tpmid2row <- data.frame(id = tpmid2row)
tpmid2row$rownum <- seq(1,nrow(tpmid2row))
head(tpmid2row)
gene2id2row <- merge(idmap,tpmid2row,by=”id”)
gene2id2row <- gene2id2row[order(gene2id2row$rownum),]
head(gene2id2row)
generownum <- gene2id2row[gene2id2row$gene==targetGene,]$rownum + 1
gtex_colnames <- read_part(“gtex_RSEM_gene_tpm.gz”, rows = 1)
gtex_tpm <- read_part(“gtex_RSEM_gene_tpm.gz”, rows = generownum)
tcga_colnames <- read_part(“tcga_RSEM_gene_tpm.gz”, rows = 1)
tcga_tpm <- read_part(“tcga_RSEM_gene_tpm.gz”, rows = generownum)
tpm <- as.numeric(c(gtex_tpm[2:length(gtex_tpm)],
tcga_tpm[2:length(tcga_tpm)] ))
names(tpm) <- c(gtex_colnames[2:length(gtex_colnames)],
tcga_colnames[2:length(tcga_colnames)] )
tcga_gtex$tpm <- tpm[tcga_gtex$sample]
#按组织排序
tcga_gtex <- arrange(tcga_gtex,tissue)
write.csv(tcga_gtex[,c(2,4,5)],”easy_input.csv”, quote = F)
tcga_gtex <- read.csv(“easy_input.csv”, row.names = 1, header = T, as.is = F)
head(tcga_gtex)
tumorlist <- unique(tcga_gtex[tcga_gtex$type2==”tumor”,]$tissue)
normallist <- unique(tcga_gtex[tcga_gtex$type2==”normal”,]$tissue)
withoutNormal <- setdiff(tumorlist, normallist)
tcga_gtex$type2 <- factor(tcga_gtex$type2,levels=c(“tumor”,”normal”))
tcga_gtex_withNormal <- tcga_gtex[!(tcga_gtex$tissue %in% withoutNormal),]
tcga_gtex_MESO <- tcga_gtex[tcga_gtex$tissue==”MESO”,]
tcga_gtex_UVM <- tcga_gtex[tcga_gtex$tissue==”UVM”,]
source(“GeomSplitViolin.R”) #位于当前文件夹
4.绘制单基因分裂小提琴图
p <- ggplot(tcga_gtex_withNormal, aes(x = tissue, y = tpm, fill = type2)) + #x对应肿瘤的类型,y对应表达量,fill填充对应组织的类型
geom_split_violin(draw_quantiles = c(0.25, 0.5, 0.75), #画4分位线
trim = F, #是否修剪小提琴图的密度曲线
linetype = “solid”, #周围线的轮廓
color = “black”, #周围线颜色
size = 0.2,
na.rm = T,
position =”identity”)+ #周围线粗细
ylab(paste0(targetGene, ” expression (TPM)”)) + xlab(“”) +
ylim(-4,9) +
scale_fill_manual(values = c(“#DF2020”, “#DDDF21”))+
theme_set(theme_set(theme_classic(base_size=20)))+
theme(axis.text.x = element_text(angle = 45, hjust = .5, vjust = .5)) + #x轴label倾斜45度
guides(fill = guide_legend(title = NULL)) +
theme(legend.background = element_blank(), #移除整体边框
#图例的左下角置于绘图区域的左下角
legend.position=c(0,0),legend.justification = c(0,0))
p + geom_split_violin(data = tcga_gtex_MESO,
mapping = aes(x = tissue, y = tpm, fill = type2),
draw_quantiles = c(0.25, 0.5, 0.75), #画4分位线
trim = F, #是否修剪小提琴图的密度曲线
linetype = “solid”, #周围线的轮廓
color = “black”, #周围线颜色
size = 0.2,
na.rm = T,
position =”identity”) +
geom_split_violin(data = tcga_gtex_UVM,
mapping = aes(x = tissue, y = tpm, fill = type2),
draw_quantiles = c(0.25, 0.5, 0.75), #画4分位线
trim = F, #是否修剪小提琴图的密度曲线
linetype = “solid”, #周围线的轮廓
color = “black”, #周围线颜色
size = 0.2,
na.rm = T,
position =”identity”) +
scale_x_discrete(limits = levels(tcga_gtex$tissue))
ggsave(paste0(targetGene, “expression.pdf”),width = 20, height = 6)
- 结果文件
TP53_expression.pdf
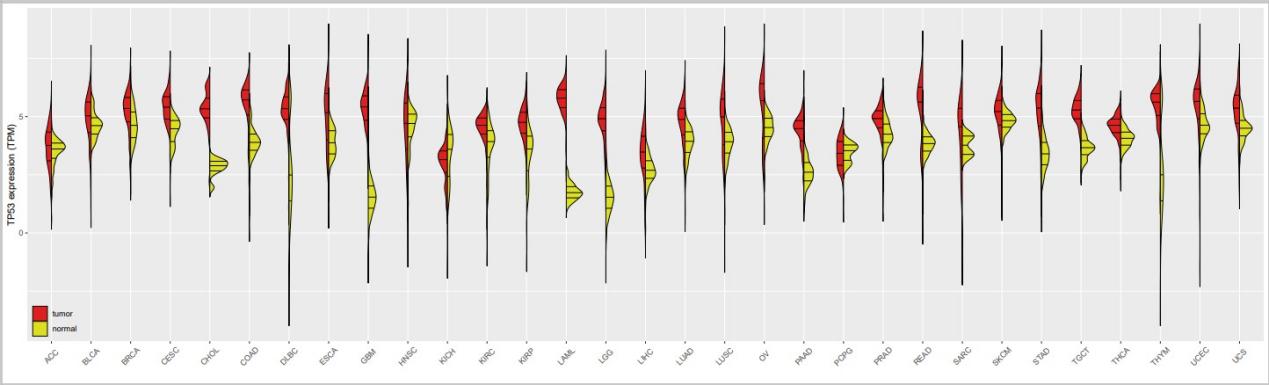
最终小果完成了单基因泛癌分裂小提琴图,看起来图片还不错哟!泛癌其他分析内容可以欢迎尝试使用本公司新开发的云平台生物信息分析小工具,零代码完成分析,云平台网址:http://www.biocloudservice.com/home.html;主要包括泛癌中体细胞拷贝数变异分析(http://www.biocloudservice.com/704/704.php),泛癌中热图和箱线图(http://www.biocloudservice.com/773/773.php),基因在肿瘤与正常组织中差异分析(http://www.biocloudservice.com/710/710.php)等相关泛癌分析小工具,小果今天的分享就到这里,欢迎大家和小果一起讨论学习奥。