小花在上篇文章说明了单基因的免疫浸润,就是对分析某个基因与免疫细胞的相关性,这种分析方法虽然不常见,但是也是在高分文章中经常见到一小部分,其实还有一种方式,小伙伴的基因太多不知道如何放,或者想把基因当作一个整体该如何分析呢?小花这里有一种方法,就是把我们基因聚类。不同的组进行分析,每个组包括相应的基因。小伙伴是不是很新奇,来看看如何去绘制吧!
原理其实很简单,小花之间教过大家一致性聚类,那么我们就用一致性聚类结果去分析,
我们先把基因聚类,这里我们需要用到一些包,没有的小伙伴去下载一下
#install.packages(“BiocManager”)
#BiocManager::install(‘ConsensusClusterPlus’)
library(tidyverse)
library(ConsensusClusterPlus)
这里示例数据还是和上一篇一样来看看:
exp <- read.table(“CESC_fpkm_mRNA_01A.txt”,sep = “\t”,row.names = 1,check.names = F,stringsAsFactors = F,header = T)
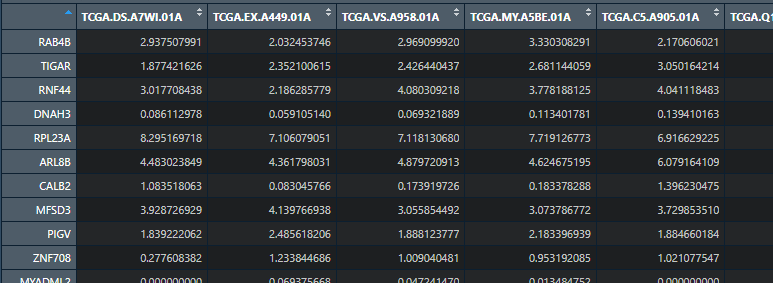
下面我们去选择要聚类的基因,并且做一些处理
d=as.matrix(exp)
gene <- c(“MMP9″,”CCND1″,”STAT1″,”AR”,”AKR1C3″,”GSTP1″,”PGR”) #这里小花随机几个基因去展示
d <- d[gene,]
mads=apply(d,1,mad)
d=d[rev(order(mads))[1:7],] #注意下这里的7是基因个数
d = sweep(d,1, apply(d,1,median,na.rm=T))
title=(“JULEI”) ##文件夹输出图片的位置,名字小伙伴可以按照自己想法去写
set.seed(1) #小花发现设不设置种子都一样
results = ConsensusClusterPlus(d,maxK=9,reps=50,pItem=0.8,pFeature=1,
title=title,clusterAlg=”hc”,distance=”pearson”,seed=1,plot=”pdf”)
results[[2]][[“consensusMatrix”]][1:5,1:5]
results[[2]][[“consensusTree”]]
results[[2]][[“consensusClass”]][1:5]
icl = calcICL(results,title=title,plot=”pdf”) ##画另一组图片
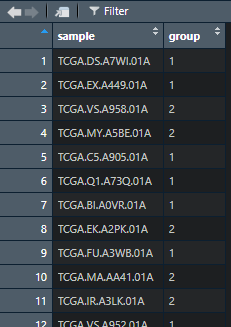
group<-results[[2]][[“consensusClass”]]
group<-as.data.frame(group)
group$group <- factor(group$group,levels=c(1,2))
save(group,file = “group_AY.Rda”)#我们保存一下聚类的分组信息,下次就是直接打开使用了
先看看结果
再看一下可视化的结果图
![]()
小花这里选择K=2的情况去展示,
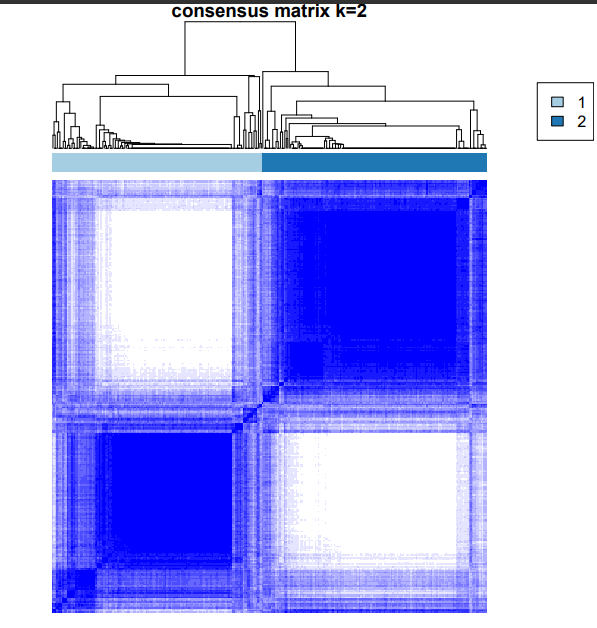
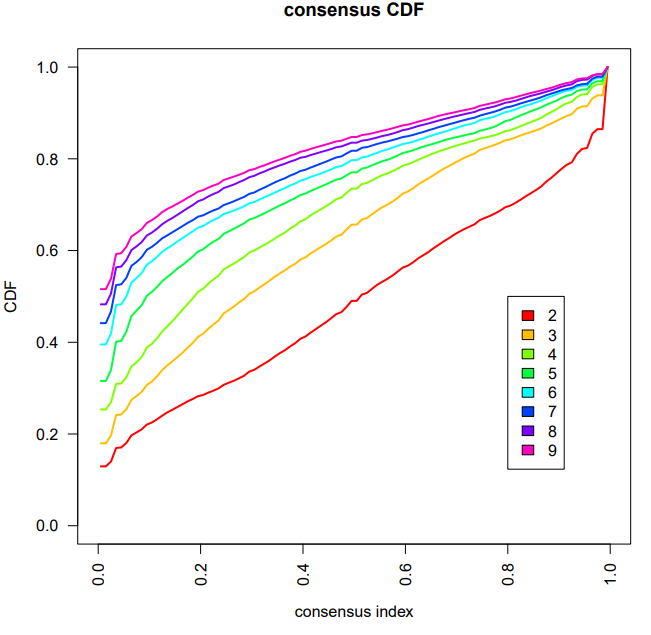
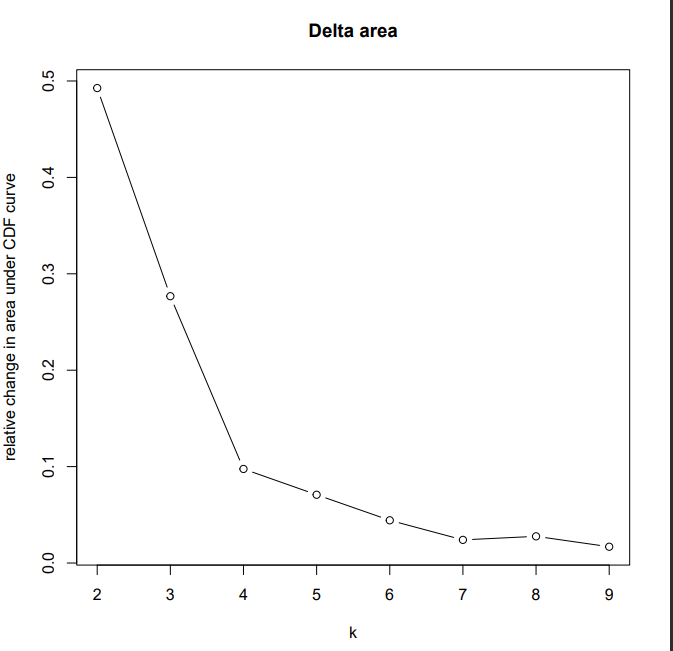
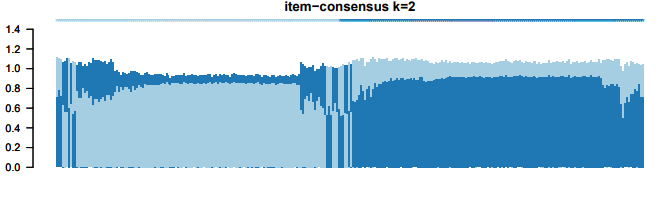
聚类结果的解读,小花这里不多赘述了,小伙伴可以看看往期小花介绍的。
这里小花设置一下配色
后续为了好看,哈哈
#配色
# 安装并加载colorspace包
#install.packages(“colorspace”)
library(colorspace)
# choosing HCL-based color palettes
# 查看所有的颜色画板
hcl_palettes(plot = TRUE)
然后我们就对聚类分组进行热图的可视化,毕竟我们要展示分组的信息的:
# 绘制ConsensusClusterPlus后的热图
library(pheatmap)
group <- group %>%
rownames_to_column(“sample”)
annotation <- group %>% arrange(group) %>% column_to_rownames(“sample”)
a <- group %>% arrange(group) %>% mutate(sample=substring(.$sample,1,12))
b <- t(exp_gene) %>%
as.data.frame() %>%
rownames_to_column(“sample”) %>%
mutate(sample=substring(.$sample,1,12))
c <- inner_join(a,b,”sample”) %>% .[,-2] %>% column_to_rownames(“sample”) %>% t(.)
pheatmap(c,annotation = annotation,
cluster_cols = F,fontsize=5,fontsize_row=5,
scale=”row”,show_colnames=F,
fontsize_col=3)
bk <- c(seq(-5,-0.1,by=0.01),seq(0,5,by=0.01))
pheatmap(c,annotation = annotation,
annotation_colors = list(group = c(“1″ =”#8B1C62”,”2″= “#EE9572″)),
cluster_cols = F,fontsize=10,fontsize_row=10,
scale=”row”,show_colnames=F,cluster_row = F,
color = c(colorRampPalette(colors = c(“#551A8B”,”white”))(length(bk)/2),colorRampPalette(colors = c(“white”,”#CD3700″))(length(bk)/2)),
#legend_breaks=seq(-3,3,1),
fontsize_col=3)
dev.off()
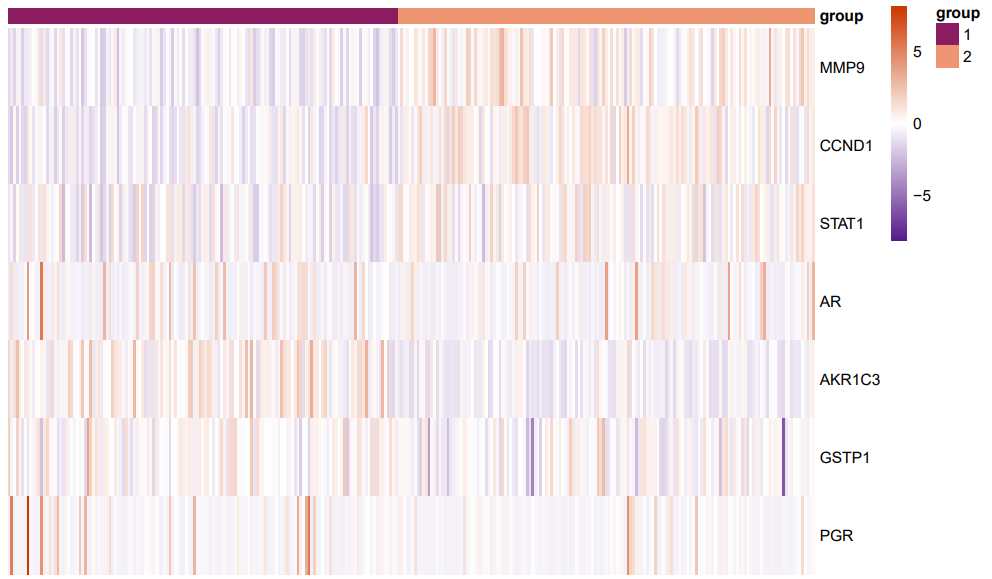
这样看来,聚类一组的高表达基因在聚类二组是低表达。那么我们就该对着两组进行免疫分析了,接下来我们进入正题,
#cluster分组CIBERSORT
setwd(“”)#可以设置一下分组
library(tidyverse)
我们这里用的免疫浸润的结果是上期小花介绍过的,小伙伴没有的可以去看上期文章,《SCI都在使用的单基因免疫浸润还不会吗?小花手把手教你学会!(一)》
a <- read.table(“CIBERSORT-Results.txt”, sep = “\t”,row.names = 1,check.names = F,header = T)
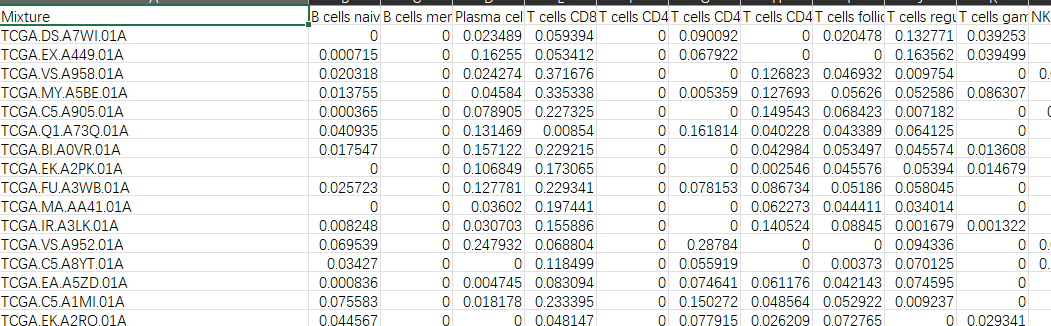
a <- a[,1:22]
identical(rownames(a),rownames(group))
b <- group
class(b$group)
a$group <- b$group
a <- a %>% rownames_to_column(“sample”)
library(ggsci)
library(tidyr)
library(ggpubr)
#install.packages(“ggsci”)
#install.packages(“tidyr”)
#install.packages(“ggpubr”)
b <- gather(a,key=CIBERSORT,value = Proportion,-c(group,sample))
ggboxplot(b, x = “CIBERSORT”, y = “Proportion”,
fill = “group”, palette = “lancet”)+
stat_compare_means(aes(group = group),
method = “wilcox.test”,
label = “p.signif”,
symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c(“***”, “**”, “*”, “ns”)))+
theme(text = element_text(size=10),
axis.text.x = element_text(angle=45, hjust=1))
dev.off()
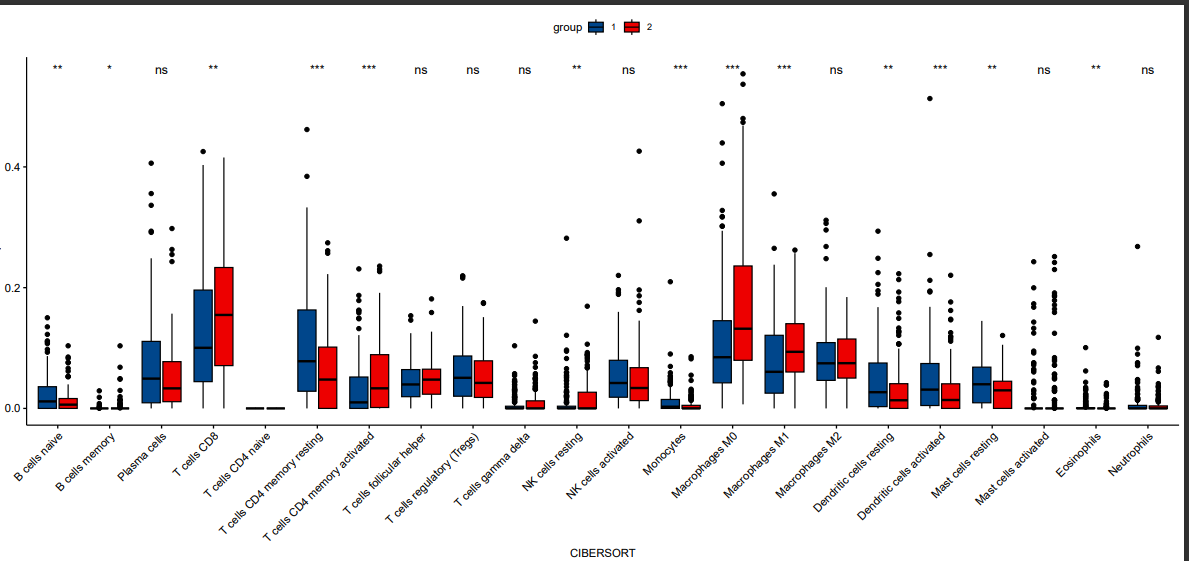
我们用小提琴图展示一下信息,这个图就是展示了不同组在不同免疫细胞中的占比,也就是正常的免疫浸润分析,
下面我们介绍最后一种免疫浸润的方式,聚类的ssgsea,之前小花专门介绍过一期ssGSEA,单样本的免疫浸润,这次我们使用聚类后的结果去绘制,来看看如何绘制的
ssGSEA
setwd(“ssGSEA_AY”)
#BiocManager::install(‘GSVA’)
library(tidyverse)
library(data.table)
#library(GSVA)
#BiocManager::install(“GSVA”)
#BiocManager::install(“DelayedArray”)
#library(DelayedArray)
library(GSVA)
#准备细胞marker
cellMarker <- data.table::fread(“cellMarker.csv”,data.table = F)
colnames(cellMarker)[2] <- “celltype”
#将cellMarker文件列名的第2个修改为celltype
type <- split(cellMarker,cellMarker$celltype)
#将cellMarker文件以celltype为分组拆分成list数据格式
#处理data.tables列表通常比使用group by参数按组对单个data.table进行操作要慢得多
cellMarker <- lapply(type, function(x){
dd = x$Metagene
unique(dd)
})
#将list中每个celltype中的基因进行合并
save(cellMarker,file = “cellMarker_ssGSEA.Rdata”)#保存中间文件
#load(“immune_infiltration//cellMarker_ssGSEA.Rdata”)
##表达量矩阵的准备
###行是基因,列是样本
expr <- data.table::fread(“CESC_fpkm_mRNA_01A.txt”,data.table = F) #读取表达文件
rownames(expr) <- expr[,1] #将第一列作为行名
expr <- expr[,-1] #去除第一列
expr <- as.matrix(expr) #将expr转换为矩阵格式
# 下面就是使用ssGSEA量化免疫浸润
gsva_data <- gsva(expr,cellMarker, method = “ssgsea”)
a <- gsva_data %>% t() %>% as.data.frame()
identical(rownames(a),rownames(group))
a$group <- group$group
a <- a %>% rownames_to_column(“sample”)
write.table(a,”ssGSEA.txt”,sep = “\t”,row.names = T,col.names = NA,quote = F)
library(ggsci)
library(tidyr)
library(ggpubr)
b <- gather(a,key=ssGSEA,value = Expression,-c(group,sample))
ggboxplot(b, x = “ssGSEA”, y = “Expression”,
fill = “group”, palette = “lancet”)+
stat_compare_means(aes(group = group),
method = “wilcox.test”,
label = “p.signif”,
symnum.args=list(cutpoints = c(0, 0.001, 0.01, 0.05, 1),
symbols = c(“***”, “**”, “*”, “ns”)))+
theme(text = element_text(size=10),
axis.text.x = element_text(angle=45, hjust=1))
dev.off() 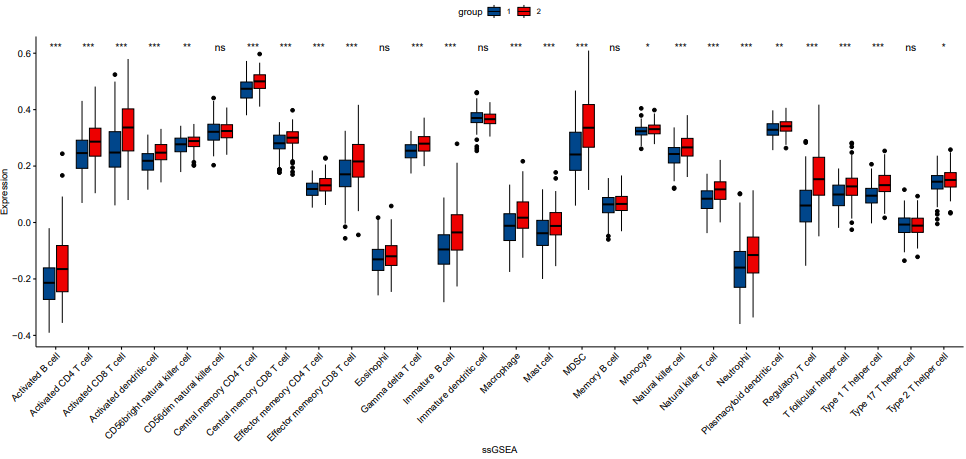
好了,到这里小花就讲解完成了,小伙伴有没有学会呢,快去拿你的数据去分析吧,但是小伙伴要理解图中的含义,不要盲目去做!