orthofinder_result(代码示例文件下载)
泛基因组可分为核心基因、附属基因、特有基因,分别指在同一物种的所有个体中出现、多个非全部个体中出现、仅一个个体中出现的基因。Orthofinder常用于做泛基因组分析,其结果内容丰富,可是如何更好地可视化,清晰看出核心基因等的分布情况呢?今天,小花来教大家使用花瓣图、饼图、upset图来对Orthofinder结果进行可视化吧。
- 首先来大概熟悉下Orthofinder的结果吧。结果包括12个文件夹,我们画图主要用到的是Orthogroups下的两个Orthogroups.GeneCount.tsv,Orthogroups_UnassignedGenes.tsv文件。


- 数据准备
GeneCount文件包含了所有同源基因组数目不为1的所有基因组,在各个样本中的数目;UnassignedGenes文件则包含了仅包含一个基因的同源基因组的样本来源信息。首先,我们要先将两个数据合并,结果文件是一个包含所有基因组的表格。
library(tidyverse)
df <- read.csv(‘data/Orthogroups.GeneCount.tsv’, row.names = 1)
df_uniq <- read.csv(‘data/Orthogroups_UnassignedGenes.tsv’, row.names = 1)
df <- df[,1:(ncol(df)-1)]
df_uniq[df_uniq != ”] <- 1
df_uniq[df_uniq == ”] <- 0
df <- rbind(df, df_uniq)

- 核心基因和特有基因统计
核心基因的筛选思路为:使用apply函数按行分析,若一行中所有值都不为0,即表示该基因组在全部样本中出现,对应的行名为核心基因。
特有基因的筛选思路为:使用apply函数按行分析,若一行中仅有1个值不为0,则为特有基因,并提取不为0列的列名为拥有该基因的个体。
附属基因:除核心基因、特有基因外者,为特有基因。
df_summary <- data.frame(gene=rownames(df))
df_summary$type <- apply(df, 1, function(x) if (length(x[x!=0]) == 1) colnames(df)[which(x != 0)] else NA)
df_summary$type[apply(df, 1, function(x) all(x != 0))] <- ‘core’
df_summary[is.na(df_summary)] <- ‘accessory’
st_u <- data.frame(t(table(df_summary$type))) %>% rename(strain=Var2) %>%select(strain, Freq) %>%
merge(., data.frame(strain = colnames(df)), all.y=T)
st_u[is.na(st_u)] <- 0
我们一起来看下整理好的数据结构,df_summary包含每个基因组类型,若为特有基因,则标记所属样本。st_u进一步统计了每个样本包含的特有基因数目。
df_summary:  table(df_summary$type):
table(df_summary$type): 
st_u: 
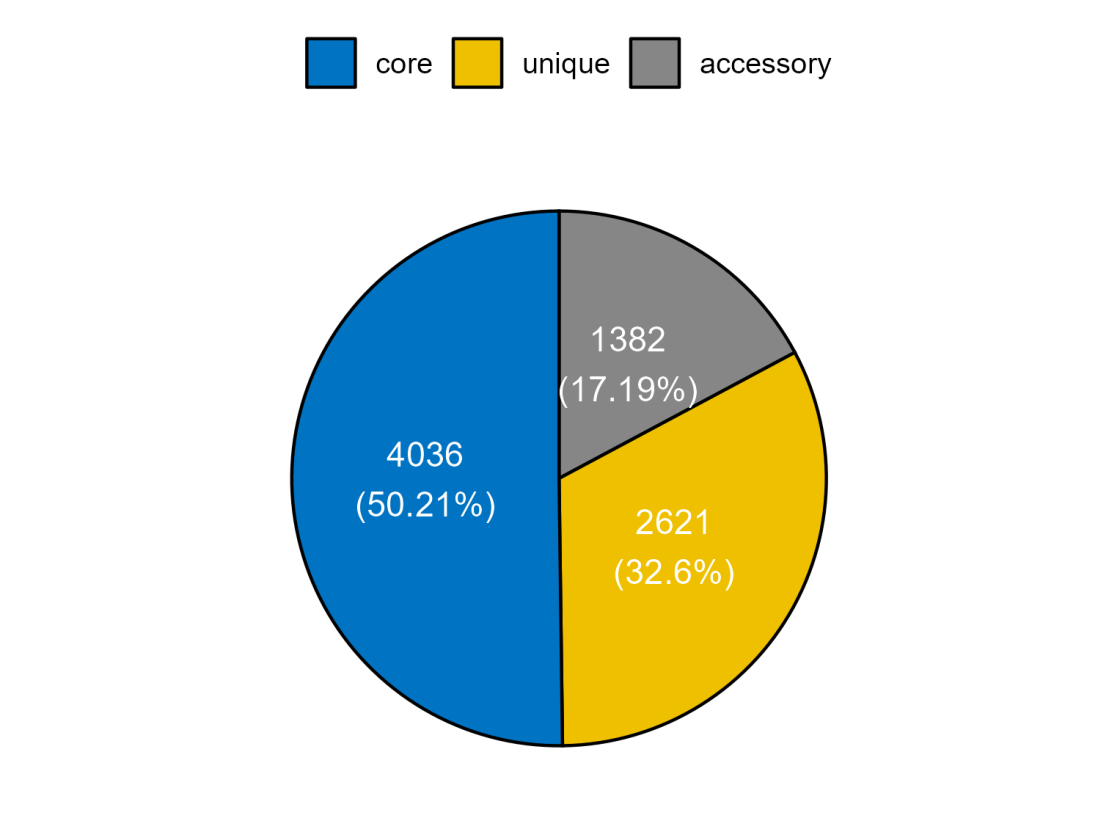
- 饼图
饼图展示核心基因、附属基因、特有基因各自的数目和比例。我们使用ggpubr来绘制。话不多说,直接上代码!
library(ggpubr)
pdata1 <- data.frame(group = factor(c(‘core’,’unique’,’accessory’), levels = c(‘core’,’unique’,’accessory’)),
freq = c(nrow(df_summary %>% filter(type==’core’)),sum(st_u$Freq), nrow(df_summary %>% filter(type==’accessory’))))
piedata1 <- pdata1%>%
mutate(labs=paste0(freq,’\n(‘,100*round(freq/sum(freq),4),’%)’))
ggpie(piedata1, ‘freq’, #绘图,只用写频数就行,切记不用再写分组
fill = ‘group’, palette = ‘jco’,#颜色板为jco.
label = ‘labs’, lab.pos = ‘in’, lab.font = c(4, ‘white’))+ #设置标签,标签的位置在图的内部,标签的大小为4, 颜色为白色.
theme(legend.position = “top”)+ labs(fill=””)
ggsave(‘result/pie.png’)
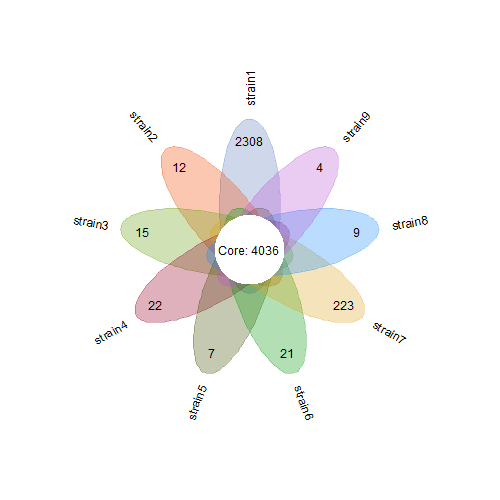
- 花瓣图
花瓣图包含了对核心基因和每个样本的特有基因的数目统计。我们基于plotrix包来完成。
首先先导入r包和绘图功能参数,并指定颜色。
需要注意的是,指定的颜色是8位字符的哦,比起6位字符颜色,最后两位表示其透明度。
此外,flower_plot <- function(){}部分,可能会有一点难懂,不过没关系,你只要把它复制粘贴运行就好啦,后面小花会教大家基础的调参哦。
ellipse_col <- c(‘#6181BD4E’,’#F348004E’,’#64A10E4E’,’#9300264E’,’#464E044E’,’#049a0b4E’,’#daa5204E’,’#1e90ff4e’,’#ba55d34E’)# 椭圆的颜色设置
flower_plot <- function(sample, otu_num, core_otu, start, a, b, r, ellipse_col, circle_col) {
par( bty = ‘n’, ann = F, xaxt = ‘n’, yaxt = ‘n’, mar = c(1,1,1,1))#start为椭圆的起始位置, a为椭圆的短轴, b为椭圆的长轴, r为中心圆的半径
plot(c(0,10),c(0,10),type=’n’)
n <- length(sample)
deg <- 360 / n
res <- lapply(1:n, function(t){
draw.ellipse(x = 5 + cos((start + deg * (t – 1)) * pi / 180), #绘制椭圆
y = 5 + sin((start + deg * (t – 1)) * pi / 180),
col = ellipse_col[t],
border = ellipse_col[t],
a = a, b = b, angle = deg * (t – 1))
text(x = 5 + 2.5 * cos((start + deg * (t – 1)) * pi / 180),#在图上设置每个样品的otu_num数目
y = 5 + 2.5 * sin((start + deg * (t – 1)) * pi / 180),
otu_num[t])
if (deg * (t – 1) < 180 && deg * (t – 1) > 0 ) {
text(x = 5 + 3.3 * cos((start + deg * (t – 1)) * pi / 180),#设置每个样品名
y = 5 + 3.3 * sin((start + deg * (t – 1)) * pi / 180),
sample[t],
srt = deg * (t – 1) – start,
adj = 1,
cex = 1
)
} else {
text(x = 5 + 3.3 * cos((start + deg * (t – 1)) * pi / 180),
y = 5 + 3.3 * sin((start + deg * (t – 1)) * pi / 180),
sample[t],
srt = deg * (t – 1) + start,
adj = 0,
cex = 1
)
}
})
draw.circle(x = 5, y = 5, r = r, col = circle_col, border = NA)#绘制中心圆
text(x = 5, y = 5, paste(‘Core:’, core_otu))#设置中心圆名字
}
接下来就是绘图啦!sample指定各个花瓣的名称,otu_num指定特有基因数目,注意要和sample对应哦,core_otu指定核心基因的数目,然后可以根据花瓣个数调整下a,b,使椭圆大小美观即可,r表示花蕊半径,ellipse_col表示花瓣颜色,也就是我们开始指定的那个,circle_col表示花蕊颜色。
png(‘result/flower.png’, w=500, h=500)
flower_plot(sample = st_u$strain, otu_num = st_u$Freq, core_otu =nrow(df_summary %>% filter(type==’core’)),
start = 90, a = 0.7, b = 2, r = 0.8, ellipse_col = ellipse_col, circle_col = ‘white’)
dev.off()
- upset图
当样本数少于5时,可以选择韦恩图表示,大于等于5时,建议使用upset图,我们需要加载upsetR包,具体使用方法小花有在《韦恩图不够用?小花教你用upset图集合可视化》一文中提到哦,这里就只简单看看效果啦。
library(UpSetR)
upsetdf <- as.data.frame(lapply(df, as.numeric))
upsetdf [upsetdf != 0] <- 1
upset(upsetdf, nset = 9,
order.by = ‘freq’)
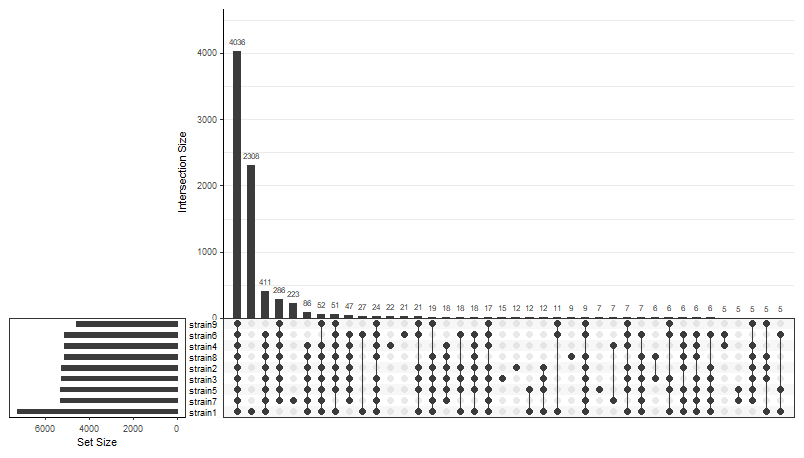
以上就是小花常用的关于orthofinder结果可视化的方法啦。饼图展示了核心基因、附属基因、特有基因的比例;花瓣图进一步展开表示了特有基因在各个样本中的分布;upset图将附属基因的样本分布也展开表示。
此外,还有其他绘图需求也欢迎范围我们的工具云生信平台在线分析http://www.biocloudservice.com/home.html。
今天的分享就到这里啦,大家还有什么好的关于orthofinder结果可视化的方法,欢迎来和小花讨论呀~