强大的基因算法工具大师:bedtools
hello,你知道有一个强大的基因组算法工具集嘛?它可以帮助我们处理基因组中生成的一系列bed、bam等格式的文件,想知道具体如何操作嘛?感兴趣的话就和小果一起看下去吧!
什么是bedtools?
Bedtools 是一个用于基因组数据分析的强大工具集,广泛应用于生物信息学和基因组学领域。它提供了一系列用于处理 BED(Browser Extensible Data)格式文件的命令行工具,可以高效地进行基因组区域操作、重叠分析、比较和注释等操作。
Bedtools 支持各种常见的基因组操作,包括并集、交集、差集、合并、排序、过滤等。通过这些操作,用户可以将不同的基因组区域文件进行组合、筛选和比较,从而实现对基因组区域的详细分析和注释。
Bedtools 还可以进行基因组区域的重叠分析。通过计算两个或多个区域文件之间的重叠情况,可以确定基因或其他功能元件在基因组上的位置关系,并进一步研究它们之间的相互作用。
除了基本的操作和重叠分析,Bedtools 还提供了一些高级功能,例如基因组区域的突变分析、统计分析以及基因组功能注释等。这使得 Bedtools 成为了生物学研究中不可或缺的工具之一。
总之,Bedtools 是一个功能强大的基因组数据处理工具集,可以帮助研究人员进行各种基因组区域的操作、分析和注释,为基因组学和生物信息学研究提供了重要的支持。
bedtools处理bed文件
bedtools在处理一系列bed文件时,根据不同的需求有不同的命令,接下来小果先为大家介绍五种最常见的操作。在这里我们先引入三个示例文件:
1.new-chr.bed
2.newArea.bed
3.newGene.bed
以拟南芥的基因组数据为基础,new-chr.bed代表拟南芥基因组上7条序列的长度区间,newArea.bed代表我们通过比对软件比对后产生的区间合集文件,newGene.bed代表我们从拟南芥基因组的注释文件中提取出的基因信息区间文件。这里的bed文件仅供说明工具的功能,大家在学习时要灵活替换成自己的文件哦!
对文件排序
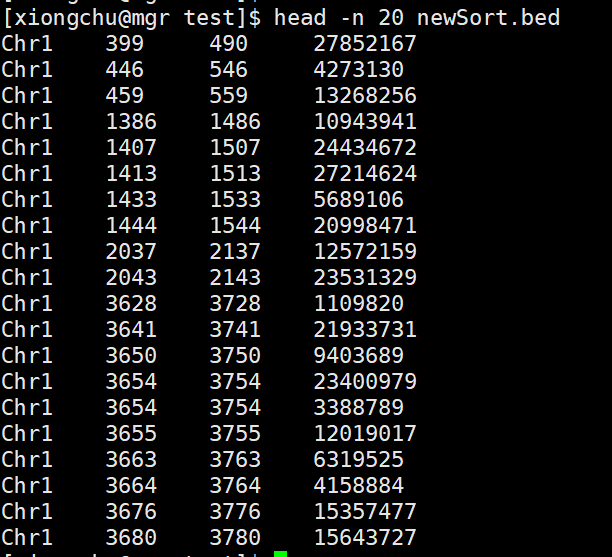
当我们拿到一个区间文件时,只有将其排序好才能保证后续的操作更加准确,所以排序这一操作往往是处理bed文件的第一步,可以使用以下命令来进行排序操作:
bedtools sort -i newArea.bed > newSort.bed
排序后,你将得到一个排序好的区间文件:
合并相同区间
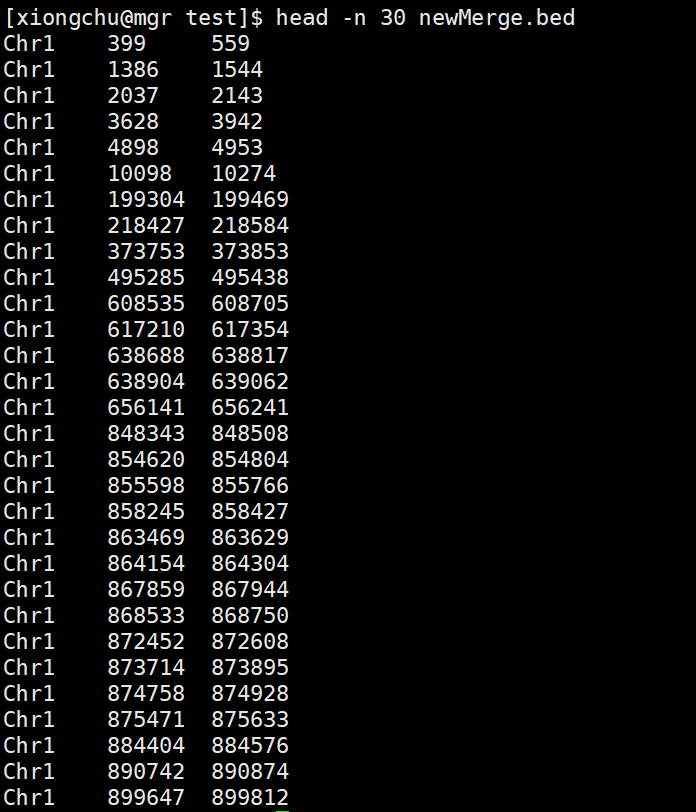
当你排序完成后,有时我们的区间中有很多“重复”的区间,合并相同以及重叠的区间可以帮助简化我们的区间,让数据更直观以及简洁,你可以使用以下命令来完成合并操作:
bedtools merge -i newSort.bed > newMerge.ned
合并后的文件格式如下:
求区间的覆盖度
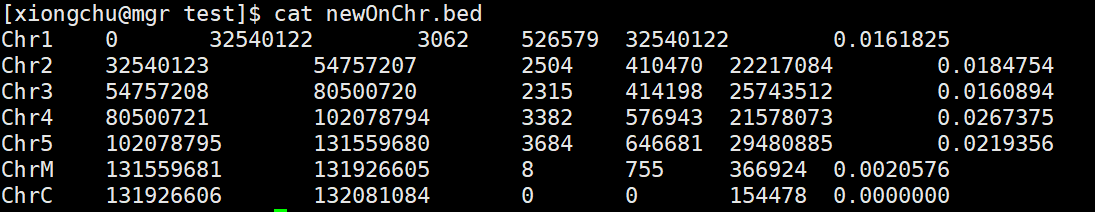
当你想求B文件在A文件上的区间覆盖度时,我们可以使用以下命令:
bedtools coverage -a A.bed -b B.bed > C.bed
eg:
bedtools coverage -a new-chr.bed -b newMerge.bed > newOnChr.bed
小果温馨提示:在这里,-a和-b后分别输入两个区间文件,其中-a后的文件参数要设置为两个区间文件中范围大的文件哦。
运行后得到的结果如下:
求两个区间的交集
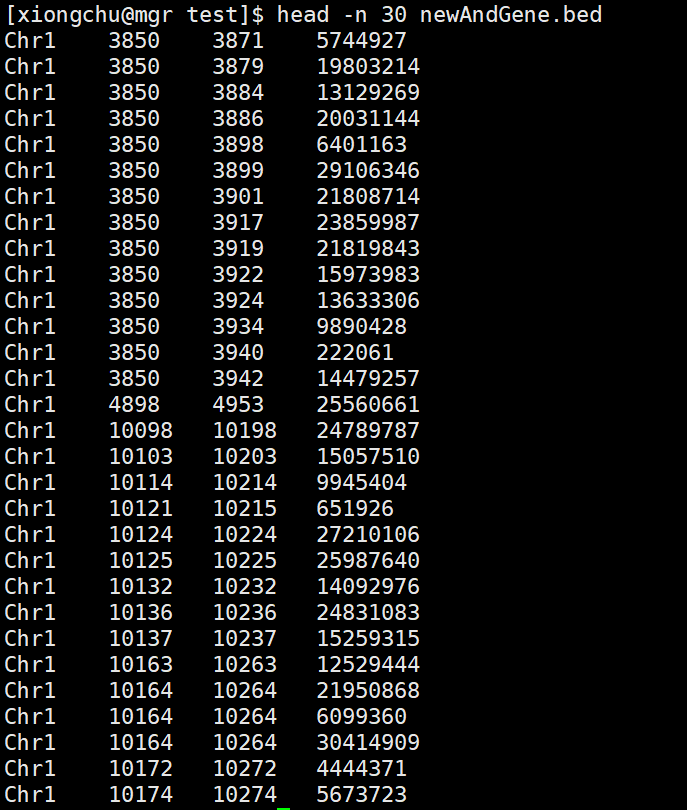
当要求两个区间文件的交集区间时,你可以使用以下命令
bedtools intersect -a A.bed -b B.bed > C.bed
eg:
bedtools intersect -a newSort.bed -b newGene.bed > newAndGene.bed
最后得到的结果如下:
求两个区间的差集
当要求两个区间的差集时,我们需要用到以下命令:
bedtools subtract -a A.bed -b B.bed > C.bed
eg:
bedtools subtract -a newSort.bed -b newGene.bed > newNoGene.bed
注意,以上命令求得的时A.bed文件中去除了B.bed文件中的区间剩余的区间文件,示例文件运行后的结果如下:
以上就是bedtools最常用的五个功能,还有很多丰富的功能等待你继续探索,怎么样,是不是很简单?赶快拿起你的基因组数据练起来吧!更多学习干货要继续关注小果哦!