背景:
关联网络分析主要用于寻找特定微生物群落在时空变化,环境过程驱动下所呈现的共现(Co-occurrence)或互斥(Co-exclusion)模式,从而分析环境差异或实验处理是否导致了群落物种装配的差异,探究微生物群落中是否存在特定的模块单元(Module)以完成特定的生态功能,并据此寻找足以撬动整个群落的组成变化的关键物种(Hubs或keystone species)。基于群落数据相关性构建的Co-occurrence网络图。可以采用R中igraph包构建并实现出图。当然,除此之外,还有一些非命令行的软件,例如cytoscape,gephi,pajek等。为了探究微生物之间的关联性,把共现性网络引入到微生物组领域,开发的ggClusterNet包可以快速的绘制你想要的网络图。
主要操作内容:
1、R环境的配置以及R包的下载
R语言下载地址https://www.r-project.org/
Rstudio下载地址:https://www.rstudio.com/products/rstudio/download/#download
R包的安装
ggClusterNet安装过程中需要很多依赖的安装包,耗时会久一些,希望你耐心等待(安装速度取决你的R环境中依赖包有多少个以前没有下载过哦~)
#安装ggClusterNet
devtools::install_github(“taowenmicro/ggClusterNet”)
2、构建数据格式
#加载所用的包
library(ggClusterNet)
library(phyloseq)
library(tidyverse)
library(igraph)
library(sna)
library(network)
#根据自己的数据格式分别导入otuable、taxnomoy和metadata数据
otutable <- read.table(“table.xls”, header = T, sep = “\t”,row.names = 1)
taxnomoy <- read.table(“tax.txt”, header = T,sep = “\t”, fill=TRUE, na.strings = “NA”, row.names = 1)
metadata <- read.table(“metadata.tsv”, header = T,sep = “\t”, row.names = 1)
#最后得到ps的数据格式
ps <- phyloseq(sample_data(metadata),otu_table(as.matrix(otutable),taxa_are_rows = T),tax_table(as.matrix(taxnomoy)))
#构建好后可以ps一下看一下这个数据集(主要包括otu_table()、sample_table()、tax_table()、phy_table()四个部分)
数据集的构建是最麻烦的地方但是一旦构建好,操作起来就很方便,希望你耐心做好这一步~
构建好数据格式后,做共现网络分析只需一行代码就可以搞定,快来试一下~
3、一键分析
#先创建一个存储的路径
path1 = “./result_net/”
dir.create(path1)
result1 = network.2(ps = ps,#构建的数据集
N = 100, #取前丰度最高的前100
layout_net = “model_Gephi.2”, #布局格式model_Gephi.2这里我用的Gephi的
r.threshold=0.6, #相关性的阈值
p.threshold=0.01, #显著性的阈值
label = TRUE, #是否在节点上标记标签
path = path1, #存放的文件
lab = “Genus”, #标签用Genus 这里一定要与taxnomoy中列名一致
zipi = TRUE #是否绘制zipi图)
4、输出文件解读
1、有两张pdf的关联网络图(networkXX.pdf),是根据包中默认的参数绘制的,但是一般图形达不到想要的理想效果。
2、会输出可以导入到Gephi中的边文件和点文件(XX_Gephi_edge.csv、XX_Gephi_node.csv),可以导入到Gephi进行调节图的布局。
5、出版级图形解读
如下图即为最后Gephi做出的图:每个节点代表一个微生物的属水平,颜色代表属所在的门类,大小代表属的相对丰度,两个属水平之间正相关用红色连接,负相关用蓝色连接。
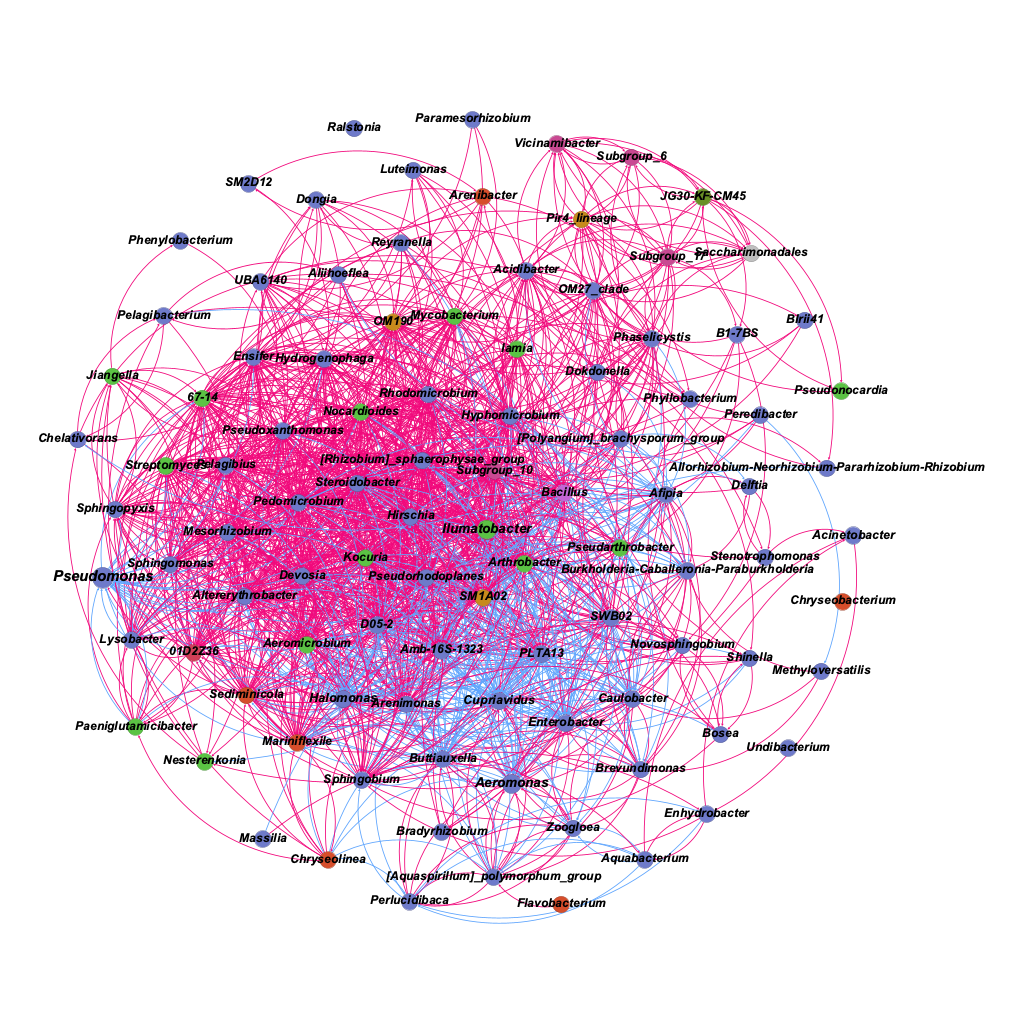
ggClusterNet的包对关联网络的布局以及一些拓扑学指数的计算都具有相应的功能,如果想绘制出满意的关联网络图,就用多多探索,这里只是简单的绘制出网络图。
好了,这样我们关联网络图就完成了。小伙伴们如果有什么问题就和小果讨论吧~