大家好呀!今天小果为大家带来了单细胞测序数据分析的干货,赶快跟随小果学起来吧!
(1)数据矩阵生成和质量控制
单细胞分析的一项关键技术进步是条形码(短的核酸片段)的发展,其允许大规模并行化,同时将成本保持在最低水平。在逆转录过程中,条形码将被添加至RNA分子中,从而可以识别单个细胞和独特的分子。
分析流程的第一步是生成数据矩阵,该矩阵通过转录物数据库从原始测序文件中呈现细胞的条形码。对于10*Genomics的数据,CellRanger是最常用的渠道,包括测序reads到基因组的解多路复用和比对,将比对的reads注释到基因,以及对基因进行量化。
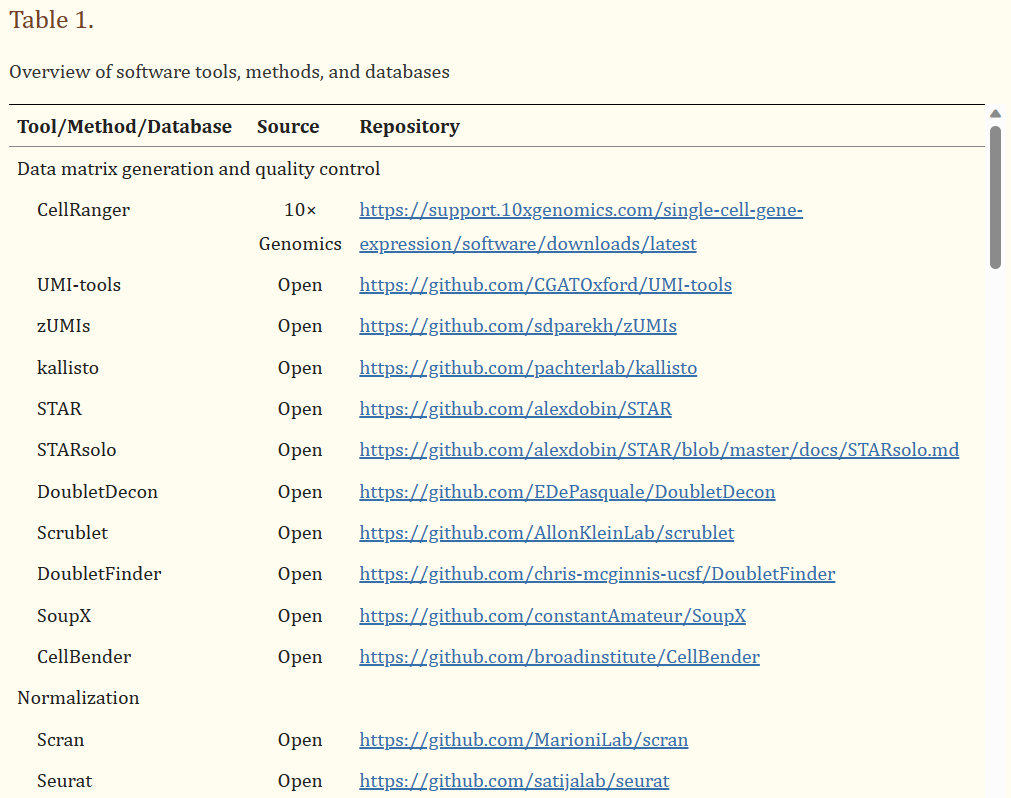
图 1 scRNA-seq分析的生物信息学工具
小果在这里仅展示部分相关工具,有关scRNA-seq分析的生物信息学工具的全面概述,请参见https://www.scrna-tools.org/tools。包括工具、方法和数据库,并带有存储库URL。、
分析流程的第二步是质量控制(QC),例如确定每个条形码的计数、基因数及其中线粒体基因的基数比例,低基因数和高线粒体读数比例通常表明细胞质量差,在这里小果推荐几个双重检测工具,包括DoubleDecon, scrulet, DoubletFinder等。
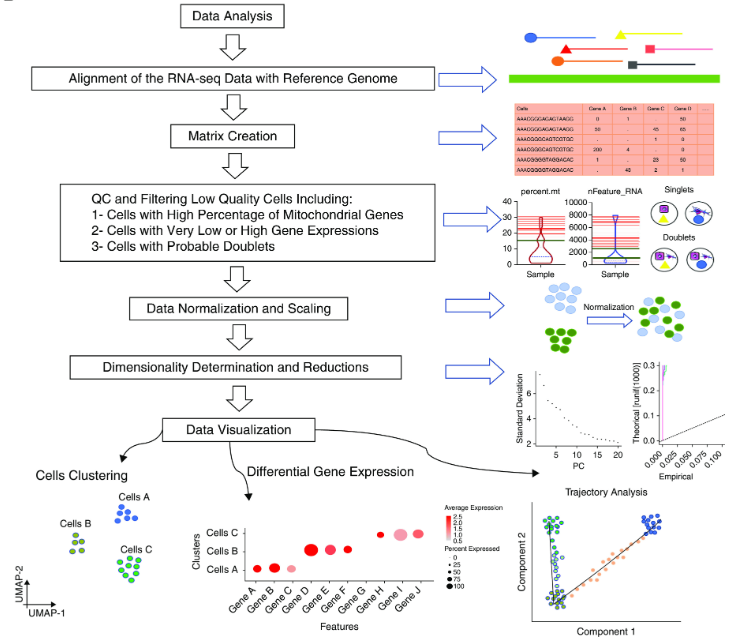
图 2 数据分析的典型步骤
(2)数据标准化
单细胞数据需要不同类型和级别的归一化,例如,总测序读取计数数改变原始计数数,因此基因计数应按比例缩放到总体计数深度。Scran使用基于池的大小因子估计和线性回归对数据进行归一化,这是除seurat使用的简单对数归一化之外最流行的方法之一。
(3)批处理效果校正和数据集成
在Seurat中,有一个基于参考的集成选项,它使用典型相关分析或互反主成分分析(PCA)最近,Harmony24越来越受欢迎,并迅速成为单细胞数据集最常用的集成方法。小果在这里推荐大家去查看Tran等人(26)和Chen等人(27)最近发表的两篇优秀论文,这两篇论文提供了一些支持Harmony、基因组实验关系关联推断和Seurat关于批效应校正的最佳证据。
(4)可视化和聚类
单细胞数据可视化主要使用其他非线性降维方法,如t分布随机邻居嵌入该方法侧重于以牺牲全局结构为代价获取局部相似性。均匀逼近和投影(UMAP)方法也因其速度快而受到欢迎UMAP似乎可以更好地捕获底层数据结构,并可以在两个以上的维度上总结数据;因此,它现在最常用于单个单元格数据可视化。聚类是一种基于距离矩阵的无监督机器学习过程。社区中默认的聚类方法是基于单个细胞k近邻的Louvain社区检测方法。单元格在图中表示为节点。每个细胞连接到它的K个最相似的细胞,这通常是使用pc减少表达空间上的欧几里得距离获得的。
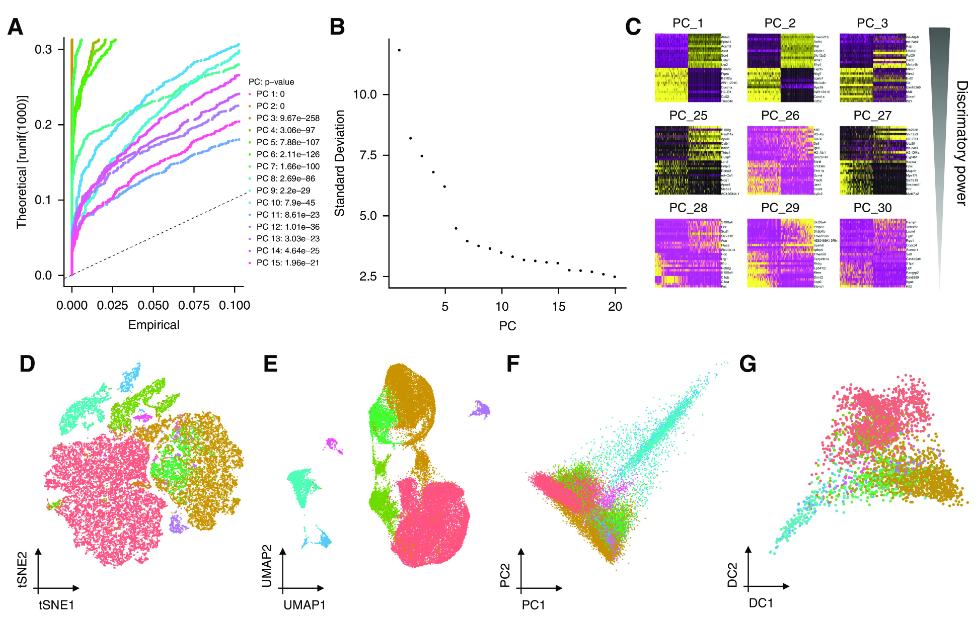
图 3 数据可视化
(5)细胞水平分析:细胞分数变化、分解和轨迹分析
Monocle是一种机器学习方法,用于重建每个细胞从一种状态过渡到另一种状态时必须执行的基因表达变化序列。小果为大家推荐一个有用的软件包是PHATE,这是一种利用数据点之间的信息几何距离捕获局部和全局非线性结构的可视化方法。推断的轨迹不一定要代表一个生物过程,应该收集进一步的证据来源来解释从这些方法中得出的轨迹。
(6)基因水平分析:差异表达、基因调控网络、驱动通路和细胞-细胞相互作用
差异表达(DE)分析通过包括技术和生物协变量对未校正的数据进行。Seurat使用不同的模型进行DE分析。MAST使用障碍模型来解释辍学为了将scRNA-seq数据集信息与其他表型变量关联起来,基于回归的模型可以将几个样本及其相关的表型特征结合起来,将某些细胞类型(如近端小管细胞)的基因表达变化与各自的定量测量表型(例如GFR、蛋白尿)关联起来。
以上便是单细胞测序数据分析的典型流程,小伙伴们有什么问题请积极和小果进行讨论哦!