生态学家用多样性和多样性来描述物种在生境内和和生境间的多样性,综合评价总体的多样性。多样性描述的是物种内的丰富度、多样性、均匀度等指标,因此也被称为生境内多样性,通常用箱线图来展现特征,用稀疏曲线样来评估测序深度。多样性指沿着环境梯度的变化,不同群落之间物种组成的差异性或是更替的速率,因此也被称为生境间多样性,通用PCoA图来展示不同样或者组本间距离。
小果以前的文章曾用过qiime2的这个软件来得到特征表和特征序列,它不仅可以解决前期的数据处理的部分,用于分析多样性的分析也是非常方便的。
α,β多样性数据分析:
qiime diversity core-metrics-phylogenetic \
–i-phylogeny rooted-tree.qza \
–i-table table.qza \
–m-metadata-file Seed-matadata.tsv \
–output-dir core-metrics-results
进化关系faith-PD、evenness数值可视化、香浓指数可视化、物种数目可视化等可视化,XXX代表打包输出的任意一种多样性指数的qza文件
qiime diversity alpha-group-significance \
–i-alpha-diversity core-metrics-results/XXX.qza \
–m-metadata-file metadata.tsv \
–o-visualization core-metrics-results/XXX.qzv
qiime diversity alpha-rarefaction \
–i-phylogeny rooted_tree.qza \
–p-metrics chao1 shannon simpson observed_otus pielou_e faith_pd goods_coverage \
–m-metadata-file map.txt
–o-visualization alpha-rarefaction
说明:–p-metrics选择不同的参数
qiime diversity beta-phylogenetic \
–i-table table_filtered.qza \
–i-phylogeny rooted-tree.qza \
–p-metric [weighted_unifrac|unweighted_unifrac] \#选择参数
–o-distance-matrix unweighted_unifrac_distance_matrix.qza#输出qza格式文件
qiime diversity beta \
–i-table table.qza \#输入OTU表
–p-metric [correlation|sokalmichener|russellrao|hamming|rogerstanimoto|chebyshev|cityblock|kulsinski|sqeuclidean|braycurtis|yule|matching|jaccard|canberra|euclidean|seuclidean|sokalsneath|wminkowski|dice|mahalanobis|cosine]\#选择参数
–o-distance-matrix unweighted_unifrac_distance_matrix.qza #输出qza格式文件
qiime diversity core-metrics \
–i-table table_filtered.qza \
–m-metadata-file map.txt
–p-sampling-depth 14208 \
–output-dir bdiv
说明:不考虑进化关系计算β多样性
以上所有输出的后缀为.qzv的格式的文件都可以在https://view.qiime2.org网站上拖动查看数据的分析结果,非常方便。如果对于图片的展示结果并不满意,可以用R进行绘图。
Aphla绘图:
一般用箱线图来展示Aphla的结果。
df <- read.table(“./Shannon.txt”,header = T,sep = “\t”)
#这里处理数据的过程可以根据实际情况而定
head(df)

p <- ggplot(df,aes(group,Chao1)) + geom_boxplot(aes(fill=group)) + theme_set(theme_bw())
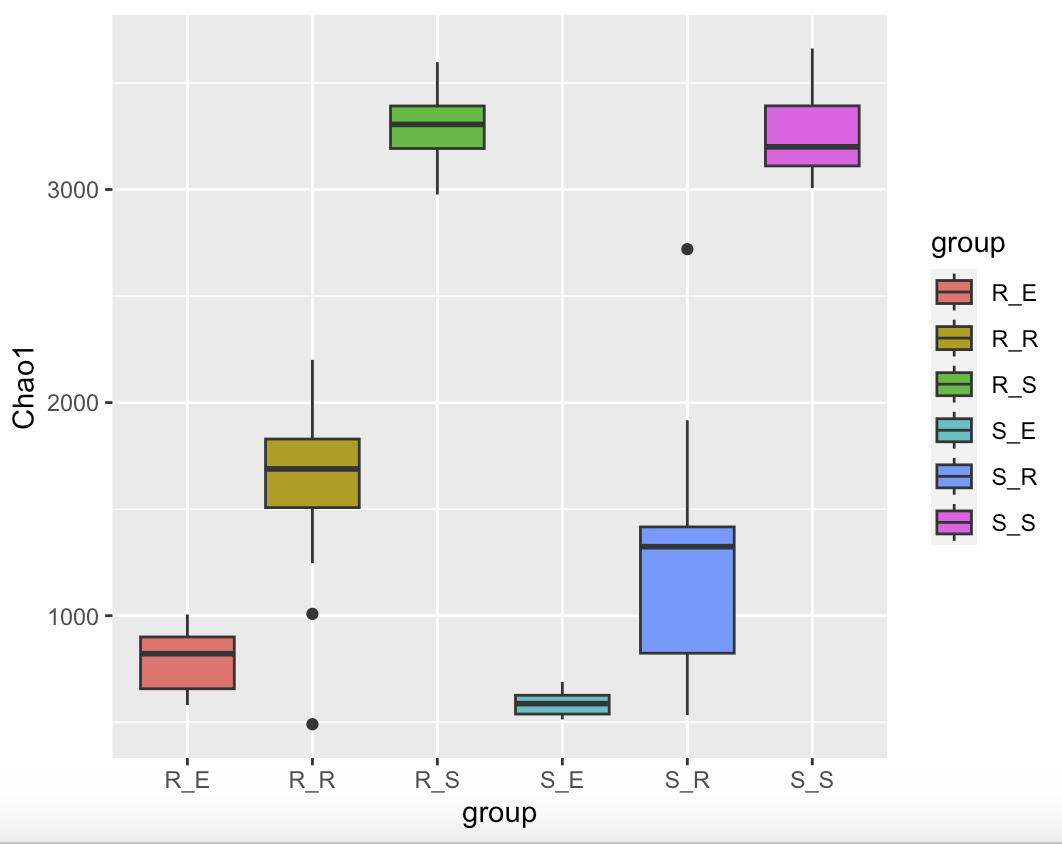
#用ggplot绘制箱线图,其它的Aphla多样性指数也是一样的绘制相应的图
Beta多样性指数
beta 多样性指数也一般用PCA、NMDS呈现,用两组检验进行验证。在小果的以前的文章中也有详细的讲解过怎样做PCA图,对于NMDS分析也可以用R中的venn包实现。具体的操作需要一篇新的文章才能讲完。
下图为PCA图和NMDS展示的Beta多样性的分析。
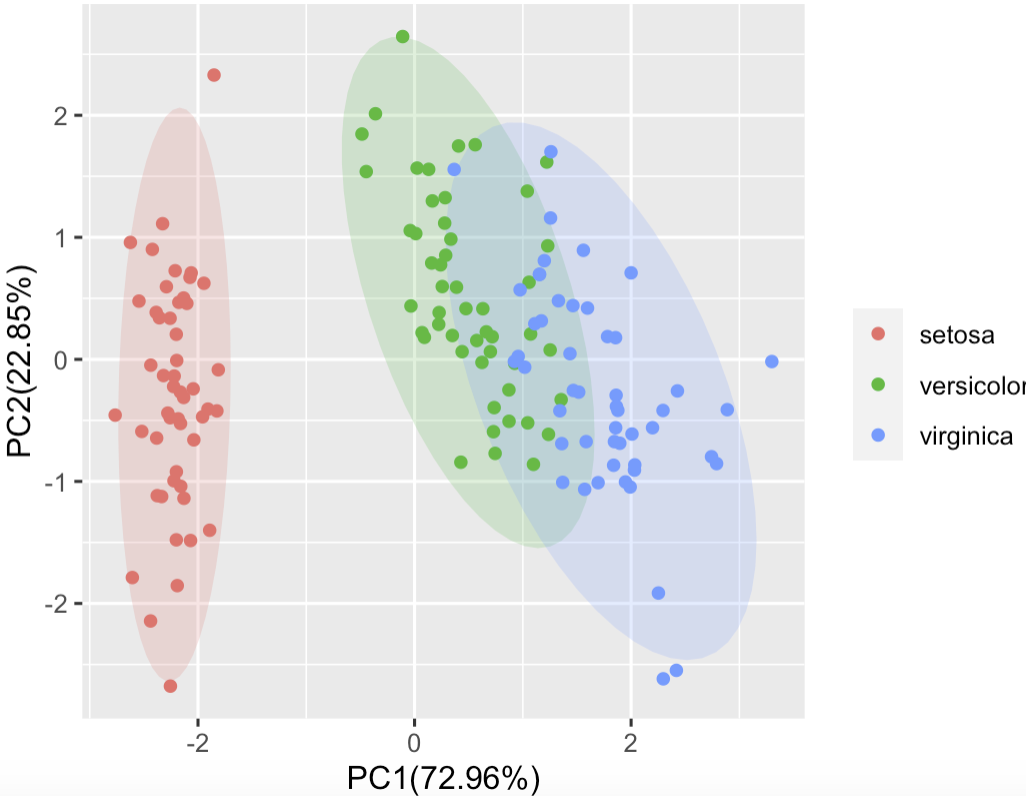
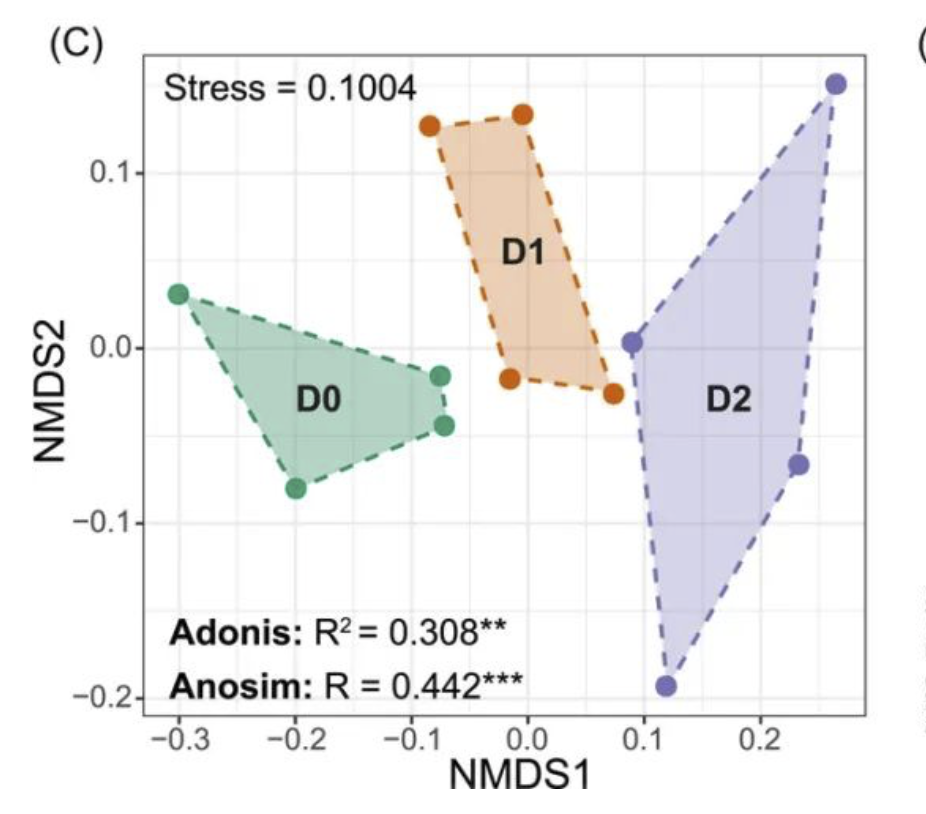
通过qiime2强大的扩增子分析平台我们不仅可以处理前期的丰度表数据,也能够利用其分析多样性分析,并能够用可视化结果。但是往往放在论文中的图片都是经过无数次的打磨的。因此我们在得到数据结果的基础上需要对数据进行更好的图形呈现。这也是无数R开发工作者在努力的目标,为科研添一点美感~
好了,这样我们微生物组的多样性就分析完成了。小伙伴们如果有什么问题就和小果讨论吧~