小果最近沉迷ATAC-seq中无法自拔,这就带大家来学习绘制ATAC-seq中的TSS富集热图。
首先,我们需要用到的软件是deeptools。这里小果给大家简单介绍一下deeptools:deeptools 是一套基于 python 开发的工具,适用于有效处理分析高通量测序数据,可用于 ChIP-seq, ATAC-seq 或 MNase-seq。deeptools 包含了多个有用的模块,可以处理 BAM 文件或 bigWig 文件,进行多种质量检查,创建标准的 bedGraph 和 bigWig 格式的归一化覆盖度文件,允许不同文件之间的比较(例如,处理组和对照组)。最后,使用这些归一化和标准化的文件,deeptools 可以创建许多适合发表的可视化图形,用于识别富集区域和进行基因组的功能注释。
简单来说,Deeptools的主要用途如下:
- 处理 bam 文件 或者 bam 转化的 bigwig 文件;
- 数据质量控制;
- 作图,比如热图、折线图
deeptools的安装十分简单,它的安装方法有多种,最简单的一种是使用 conda 命令:
conda install -c bioconda deeptools
这样可以自动安装 deeptools 及其所有的 python 依赖项
另一种方法是使用 pip 命令:
pip install deeptools
这样也可以自动安装 deeptools 及其所有的 python 依赖项
这里小果考考小伙伴们,你们还记得TSS是什么吗?如果你知道的话,那么小果觉得你真是泰酷辣!
揭晓答案咯:
TSS 是转录起始位点(transcription start site)的缩写,是指一个基因的 5’ 端转录的第一个碱基,它是与新生 RNA 链第一个核苷酸相对应 DNA 链上的碱基,通常为一个嘌呤(A 或 G)。TSS 是基因转录的起点,也是启动子的一部分。启动子是一段区域,与 RNA 聚合酶结合并能够起始 mRNA 的合成。TSS 前即 5’ 末端的序列称为上游,而把其后即 3’ 末端的序列称为下游。
Deeptools下载完成之后,小果开始带小伙伴们画TSS富集图咯
首先准备bw文件,先建索引,然后转bw,然后取注释bed文件,接着形成矩阵文件,最后绘制TSS富集图
下面是第一步:对bam文件建立索引,转bw文件
samtools index test.bam
bamCoverage -b test.bam -o test.bw
下面是获取注释bed文件,这里需要用到选用物种的参考基因组注释文件嗷
awk ‘$3 == “gene”‘ test.gff3 | awk ‘BEGIN{FS=”\t|=|;”;OFS=”\t”}{print $1,$4-1,$4}’ > gene.bed
又用到了我们的老朋友awk,不会的小伙伴们赶紧学起来呀
下面是形成矩阵文件,用 ComputeMatrix 计算全基因组范围内 peaks 在基因特征的分布情况。
小果的代码是这样的:
computeMatrix reference-point –referencePoint TSS -p 15 -b 10000 -a 10000 -R gene.bed -S test.bw –skipZeros -o test_TSS.gz –outFileSortedRegions test_genes.bed
下面是参数的解释:
reference-point # 选择模式
-p 15 # 线程数
–referencePoint TSS # 选择参考点,还可以选择TES, center
-b 10000 -a 10000 # 感兴趣的区域,-b上游,-a下游
-R # 基因注释信息
-S # 提供的 bigwig 文件
–skipZeros
–outFileSortedRegions # 输出的文件名
下面就是最后一步绘制富集图啦,小果的代码是这样的:
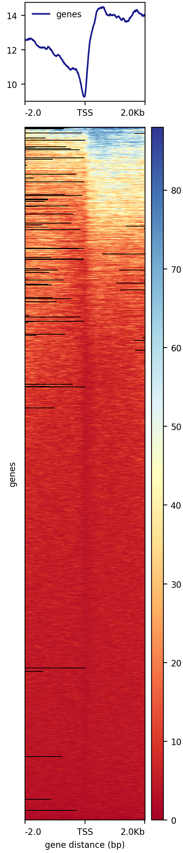
plotHeatmap -m test_TSS.gz -out test_Heatmap.png
下面就是小果画出来的TSS富集图啦,是不是很美观呢?
今天的TSS富集热图学习就到这里啦,感兴趣的小伙伴可以找小果讨论哦,我们明天见咯~