今天小果给小伙伴带来的分享是啥呢?今天小果将复现一篇8+生信文章中的WGCNA分析,通过该推文将掌握如何利用自己的数据进行WGCNA分析,来进行与表型或者疾病相关候选基因的挖掘,小果觉得WGCNA是一个非常不错的候选基因挖掘的方法,值得小伙伴们认真学习,接下来跟着小果开始今天的分享吧!
1.WGCNA介绍
在开始分析之前,小果想为小伙伴简单介绍一下WGCNA,WGCNA是一种用于基因共表达的方法,它可以将高通量基因表达数据转化为一个共表达网络,从而发现具有相似表达模式的基因模块,以及研究这些模块与表型之间的相关性,最终挖掘与表型相关的候选基因。这就是小果对WGCNA分析原理的简单介绍,是不是通俗易懂奥!其实WGCNA分析很简单,小伙伴不要因为分析步骤多而不敢尝试,只需要输入基因表达矩阵和对应样本表型数据文件,就可以完成分析,有需要的小伙伴可以跟着小果开始今天的实操。
2.准备需要的R包
#安装需要的R包
BiocManager::install(“WGCNA”)
#加载需要的R包
library(WGCNA)
3.WGCNA分析
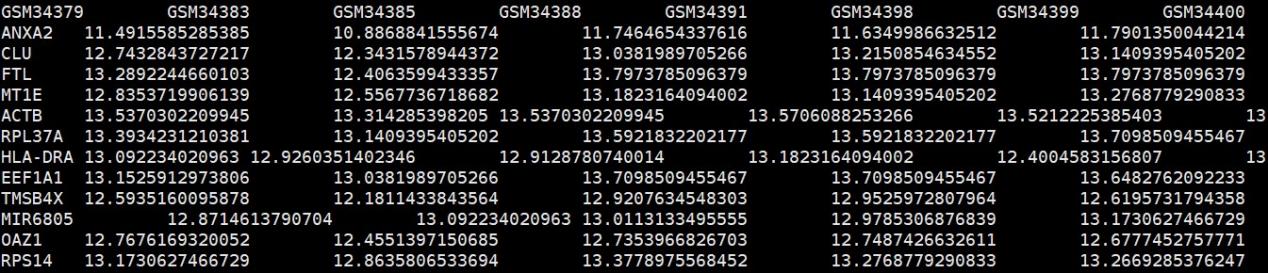
#读取表达矩阵,行名为基因,列名为样本信息
expr<-read.table(“combined.expr.txt”,header=T,sep=”\t”)
#行列转置
mydata<-expr
expr2<-data.frame(t(mydata))
#确定expr2列名
colnames(expr2)<-rownames(mydata)
#确定expr2行名
rownames(expr2)<-colnames(mydata)
#基因过滤
dataExpr1<-expr2
gsg=goodSamplesGenes(dataExpr1,verbose=3);
gsg$allOK
if (!gsg$allOK){
# Optionally, print the gene and sample names that were removed:
if (sum(!gsg$goodGenes)>0)
printFlush(paste(“Removing genes:”,
paste(names(dataExpr)[!gsg$goodGenes], collapse = “,”)));
if (sum(!gsg$goodSamples)>0)
printFlush(paste(“Removing samples:”,
paste(rownames(dataExpr)[!gsg$goodSamples], collapse = “,”)));
# Remove the offending genes and samples from the data:
dataExpr1 = dataExpr1[gsg$goodSamples, gsg$goodGenes]
}
#基因数目
nGenes = ncol(dataExpr1)
#样本数目
nSamples = nrow(dataExpr1)
#导入表型数据,第一列为样本信息,其他列为表型数据,需要注意的是表型数据要与样本数据一一对应
traitData=read.table(“TraitData.txt”,header=T,sep=”\t”,row.names=1)

#提取表达矩阵样本名
fpkmSamples<-rownames(dataExpr1)
#提取表型文件样本名
traitSamples<-rownames(traitData)
#表达矩阵样本名顺序匹配表型文件样本名
traitRows<-match(fpkmSamples,traitSamples)
#获得匹配好样本顺序的表型文件
dataTraits<-traitData[traitRows,]
#绘制树+表型热图
sampleTree2<-hclust(dist(dataExpr1),method=”average”)
traitColors<-numbers2colors(dataTraits,signed=FALSE)
png(“sample-subtype-cluster.png”,width = 800,height = 600)
plotDendroAndColors(sampleTree2,traitColors,groupLabels=names(dataTraits),main=”Sample dendrogram and trait heatmap”,
cex.colorLabels=1.5,cex.dendroLabels=1,cex.rowText=2)
dev.off()

#计算合适的power值,并绘制Power值曲线图
powers = c(c(1:10), seq(from = 12, to=20, by=2))
sft = pickSoftThreshold(dataExpr1, powerVector = powers, verbose = 5)
#设置网络构建参数选择范围,计算无尺度分布拓扑矩阵
png(“step2-beta-value.png”,width = 800,height = 600)
par(mfrow = c(1,2));
cex1 = 0.9;
# Scale-free topology fit index as a function of the soft-thresholding power
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab=”Soft Threshold (power)”,ylab=”Scale Free Topology Model Fit,signed R^2″,type=”n”,
main = paste(“Scale independence”));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col=”red”);
# this line corresponds to using an R^2 cut-off of h
abline(h=0.90,col=”red”)
# Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab=”Soft Threshold (power)”,ylab=”Mean Connectivity”, type=”n”,
main = paste(“Mean connectivity”))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col=”red”)
dev.off()

#一步法构建共表达网络
##需要调用WGCNA包自带的cor函数,不然会发生报错奥!
cor<-WGCNA::cor
##在进行共表达网络构建时,power值的选择非常重要,最影响结果的一个参数,需要经过多次尝试,才能找到最适合的。
net = blockwiseModules(dataExpr1, power = 7, maxBlockSize = nGenes,
TOMType =’unsigned’, minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs=TRUE, saveTOMFileBase = “drought”,
verbose = 3)
table(net$colors)
cor<-stats::cor
#绘制基因聚类树和模块颜色组合
# Convert labels to colors for plotting
moduleLabels = net$colors
moduleColors = labels2colors(moduleLabels)
# Plot the dendrogram and the module colors underneath
png(“step4-genes-modules.png”,width = 800,height = 600)
plotDendroAndColors(net$dendrograms[[1]], moduleColors[net$blockGenes[[1]]],
“Module colors”,
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
dev.off()

#计算模块与性状间的相关性及绘制相关性热图
nGenes = ncol(dataExpr1)
nSamples = nrow(dataExpr1)
design=read.table(“TraitData.txt”, sep = ‘\t’, header = T, row.names = 1)
# Recalculate MEs with color labels
MEs0 = moduleEigengenes(dataExpr1, moduleColors)$eigengenes
##不同颜色的模块的ME值矩 (样本vs模块)
MEs = orderMEs(MEs0);
moduleTraitCor = cor(MEs, design , use = “p”);
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples)
# Will display correlations and their p-values
textMatrix = paste(signif(moduleTraitCor, 2), “\n(“,
signif(moduleTraitPvalue, 1), “)”, sep = “”);
dim(textMatrix) = dim(moduleTraitCor)
png(“step5-Module-trait-relationships.png”,width = 800,height = 1200,res = 120)
par(mar = c(6, 8.5, 3, 3));
# Display the correlation values within a heatmap plot
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = colnames(design),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste(“Module-trait relationships”))
dev.off()

#计算MM值和GS值并绘图
##切割,从第三个字符开始保存
modNames = substring(names(MEs), 3)
geneModuleMembership = as.data.frame(cor(dataExpr1, MEs, use = “p”));
## 算出每个模块跟基因的皮尔森相关系数矩
## MEs是每个模块在每个样本里面皿
## dataExpr1是每个基因在每个样本的表达量
MMPvalue = as.data.frame(
corPvalueStudent(as.matrix(geneModuleMembership), nSamples) ##计算MM值对应的P值
);
names(geneModuleMembership) = paste(“MM”, modNames, sep=””); ##给MM对象统一赋名
names(MMPvalue) = paste(“p.MM”, modNames, sep=””); ##给MMPvalue对象统一赋名
##计算基因与每个性状的显著性(相关性)及pvalue值
geneTraitSignificance = as.data.frame(cor(dataExpr1, design, use = “p”));
GSPvalue = as.data.frame(
corPvalueStudent(as.matrix(geneTraitSignificance), nSamples) ##计算GS值对应的pvalue值
);
names(geneTraitSignificance) = paste(“GS.”, colnames(design), sep=””);
names(GSPvalue) = paste(“p.GS.”, colnames(design), sep=””);
module = “brown”
column = match(module, modNames); ##在所有模块中匹配选择的模块,返回所在的位置
moduleGenes = moduleColors==module; ##==匹配,匹配上的返回True,否则返回false
png(“step6-Module_membership-gene_significance.png”,width = 800,height = 600)
par(mfrow = c(1,1)); ##设定输出图片行列数量,(1_1)代表行一个,列一丿
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]), ## 绘制MM和GS散点囿
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste(“Module Membership in”, module, “module”),
ylab = “Gene significance for Basal”,
main = paste(“Module membership vs. gene significance\n”),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)
dev.off()

#绘制TOM热图+模块性状组合图
geneTree = net$dendrograms[[1]]; ##提取基因聚类
dissTOM = 1-TOMsimilarityFromExpr(dataExpr1, power = 7); ##计算TOM距离
plotTOM = dissTOM^7; ##通过7次方处理进行标准化,降低基因间的误差
diag(plotTOM) = NA; ##替换斜对角矩阵中的内容为NA
TOMplot(plotTOM, geneTree, moduleColors, main = “Network heatmap plot, all genes”)
nSelect = 400 ##挑选需要提取的基因
set.seed(10); ##设置随机种子数,保证选取的随机性
select = sample(nGenes, size = nSelect); ##仿5000个基因中随机叿400丿
selectTOM = dissTOM[select, select]; ##提取挑出来的基因的TOM距离
# There’s no simple way of restricting a clustering tree to a subset of genes, so we must re-cluster.
selectTree = hclust(as.dist(selectTOM), method = “average”) ##对挑出来对基因TOM距离重新聚类
selectColors = moduleColors[select];
# Open a graphical window
sizeGrWindow(9,9)
# Taking the dissimilarity to a power, say 10, makes the plot more informative by effectively changing
# the color palette; setting the diagonal to NA also improves the clarity of the plot
plotDiss = selectTOM^7; ##对挑选出对基因TOM距离7次方处理
diag(plotDiss) = NA; ##替换斜对角矩阵中的内容为NA
png(“step7-Network-heatmap.png”,width = 800,height = 600)
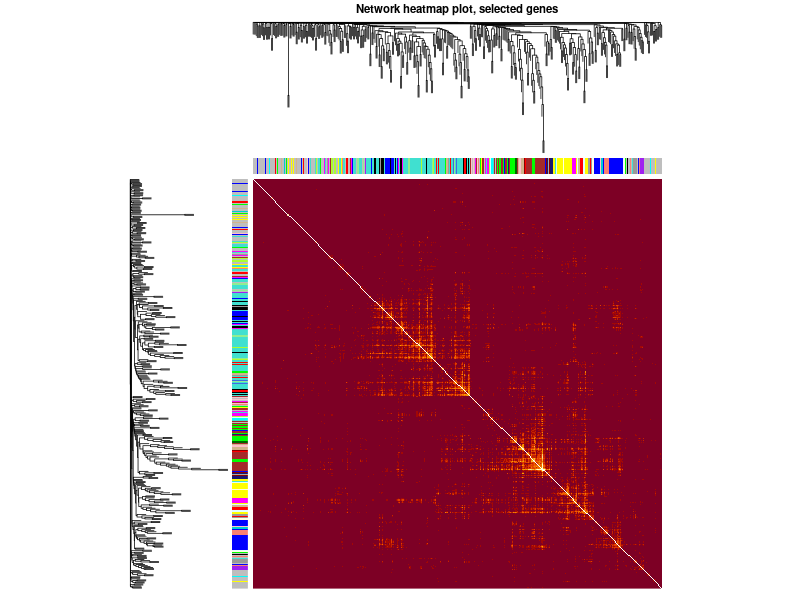
TOMplot(plotDiss, selectTree, selectColors, ##绘制TOM图
main = “Network heatmap plot, selected genes”)
dev.off()
# 模块性状组合图
RA = as.data.frame(design[,2]); ##提取特定性状
names(RA) = “RA”
# Add the weight to existing module eigengenes
MET = orderMEs(cbind(MEs, RA))
# Plot the relationships among the eigengenes and the trait
sizeGrWindow(5,7.5);
par(cex = 0.9)
png(“step7-Eigengene-dendrogram.png”,width = 800,height = 600)
plotEigengeneNetworks(MET, “”, ##绘制模块聚类图和热图
marDendro =c(0,5,1,5), ##树类图区间大小谁宿
marHeatmap = c(5,6,1,2), cex.lab = 0.8, ##树类图区间大小谁宿
xLabelsAngle = 90) ##轴鲜艿90庿
dev.off()

#导出单个模块的网络结果
TOM = TOMsimilarityFromExpr(dataExpr1, power = 7); ##需要较长时间
# 选择brown模块
module = “brown”;
# Select module probes
probes = colnames(dataExpr1)
inModule = (moduleColors==module);
## 提取指定模块的基因名
modProbes = probes[inModule];

modTOM = TOM[inModule, inModule];
dimnames(modTOM) = list(modProbes, modProbes)
## 模块对应的基因关系矩
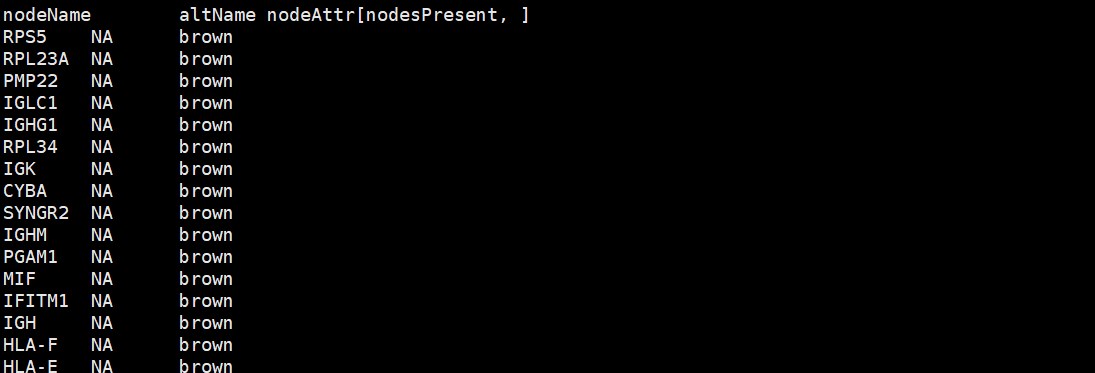
cyt = exportNetworkToCytoscape( ##导出单个模块的Cytoscape输入文件
modTOM,
edgeFile = paste(“CytoscapeInput-edges-“, paste(module, collapse=”-“), “.txt”, sep=””), ##输出边文件名
nodeFile = paste(“CytoscapeInput-nodes-“, paste(module, collapse=”-“), “.txt”, sep=””), ##输出点文件名
weighted = TRUE,
threshold = 0.02,
nodeNames = modProbes,
nodeAttr = moduleColors[inModule]
)
CytoscapeInput-edges-brown.txt,边文件

CytoscapeInput-nodes-brown.txt,点文件
最终小果成功完成了WGCNA分析,基本达到了复现,今天小果的分享就到这里啦!如果小伙伴有其他数据分析需求,可以尝试本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:http://www.biocloudservice.com/home.html,包括了WGCNA分析(http://www.biocloudservice.com/336/336.php),GEO数据下载(http://www.biocloudservice.com/371/371.php)等小工具,欢迎大家和小果一起讨论学习哈!!!!
高分生信SCI-WGCNA分析复现
今天小果给小伙伴带来的分享是啥呢?今天小果将复现一篇8+生信文章中的WGCNA分析,通过该推文将掌握如何利用自己的数据进行WGCNA分析,来进行与表型或者疾病相关候选基因的挖掘,小果觉得WGCNA是一个非常不错的候选基因挖掘的方法,值得小伙伴们认真学习,接下来跟着小果开始今天的分享吧!
1.WGCNA介绍
在开始分析之前,小果想为小伙伴简单介绍一下WGCNA,WGCNA是一种用于基因共表达的方法,它可以将高通量基因表达数据转化为一个共表达网络,从而发现具有相似表达模式的基因模块,以及研究这些模块与表型之间的相关性,最终挖掘与表型相关的候选基因。这就是小果对WGCNA分析原理的简单介绍,是不是通俗易懂奥!其实WGCNA分析很简单,小伙伴不要因为分析步骤多而不敢尝试,只需要输入基因表达矩阵和对应样本表型数据文件,就可以完成分析,有需要的小伙伴可以跟着小果开始今天的实操。
2.准备需要的R包
#安装需要的R包
BiocManager::install(“WGCNA”)
#加载需要的R包
library(WGCNA)
3.WGCNA分析
#读取表达矩阵,行名为基因,列名为样本信息
expr<-read.table(“combined.expr.txt”,header=T,sep=”\t”)
#行列转置
mydata<-expr
expr2<-data.frame(t(mydata))
#确定expr2列名
colnames(expr2)<-rownames(mydata)
#确定expr2行名
rownames(expr2)<-colnames(mydata)
#基因过滤
dataExpr1<-expr2
gsg=goodSamplesGenes(dataExpr1,verbose=3);
gsg$allOK
if (!gsg$allOK){
# Optionally, print the gene and sample names that were removed:
if (sum(!gsg$goodGenes)>0)
printFlush(paste(“Removing genes:”,
paste(names(dataExpr)[!gsg$goodGenes], collapse = “,”)));
if (sum(!gsg$goodSamples)>0)
printFlush(paste(“Removing samples:”,
paste(rownames(dataExpr)[!gsg$goodSamples], collapse = “,”)));
# Remove the offending genes and samples from the data:
dataExpr1 = dataExpr1[gsg$goodSamples, gsg$goodGenes]
}
#基因数目
nGenes = ncol(dataExpr1)
#样本数目
nSamples = nrow(dataExpr1)
#导入表型数据,第一列为样本信息,其他列为表型数据,需要注意的是表型数据要与样本数据一一对应
traitData=read.table(“TraitData.txt”,header=T,sep=”\t”,row.names=1)
#提取表达矩阵样本名
fpkmSamples<-rownames(dataExpr1)
#提取表型文件样本名
traitSamples<-rownames(traitData)
#表达矩阵样本名顺序匹配表型文件样本名
traitRows<-match(fpkmSamples,traitSamples)
#获得匹配好样本顺序的表型文件
dataTraits<-traitData[traitRows,]
#绘制树+表型热图
sampleTree2<-hclust(dist(dataExpr1),method=”average”)
traitColors<-numbers2colors(dataTraits,signed=FALSE)
png(“sample-subtype-cluster.png”,width = 800,height = 600)
plotDendroAndColors(sampleTree2,traitColors,groupLabels=names(dataTraits),main=”Sample dendrogram and trait heatmap”,
cex.colorLabels=1.5,cex.dendroLabels=1,cex.rowText=2)
dev.off()
#计算合适的power值,并绘制Power值曲线图
powers = c(c(1:10), seq(from = 12, to=20, by=2))
sft = pickSoftThreshold(dataExpr1, powerVector = powers, verbose = 5)
#设置网络构建参数选择范围,计算无尺度分布拓扑矩阵
png(“step2-beta-value.png”,width = 800,height = 600)
par(mfrow = c(1,2));
cex1 = 0.9;
# Scale-free topology fit index as a function of the soft-thresholding power
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab=”Soft Threshold (power)”,ylab=”Scale Free Topology Model Fit,signed R^2″,type=”n”,
main = paste(“Scale independence”));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col=”red”);
# this line corresponds to using an R^2 cut-off of h
abline(h=0.90,col=”red”)
# Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab=”Soft Threshold (power)”,ylab=”Mean Connectivity”, type=”n”,
main = paste(“Mean connectivity”))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col=”red”)
dev.off()
#一步法构建共表达网络
##需要调用WGCNA包自带的cor函数,不然会发生报错奥!
cor<-WGCNA::cor
##在进行共表达网络构建时,power值的选择非常重要,最影响结果的一个参数,需要经过多次尝试,才能找到最适合的。
net = blockwiseModules(dataExpr1, power = 7, maxBlockSize = nGenes,
TOMType =’unsigned’, minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs=TRUE, saveTOMFileBase = “drought”,
verbose = 3)
table(net$colors)
cor<-stats::cor
#绘制基因聚类树和模块颜色组合
# Convert labels to colors for plotting
moduleLabels = net$colors
moduleColors = labels2colors(moduleLabels)
# Plot the dendrogram and the module colors underneath
png(“step4-genes-modules.png”,width = 800,height = 600)
plotDendroAndColors(net$dendrograms[[1]], moduleColors[net$blockGenes[[1]]],
“Module colors”,
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
dev.off()
#计算模块与性状间的相关性及绘制相关性热图
nGenes = ncol(dataExpr1)
nSamples = nrow(dataExpr1)
design=read.table(“TraitData.txt”, sep = ‘\t’, header = T, row.names = 1)
# Recalculate MEs with color labels
MEs0 = moduleEigengenes(dataExpr1, moduleColors)$eigengenes
##不同颜色的模块的ME值矩 (样本vs模块)
MEs = orderMEs(MEs0);
moduleTraitCor = cor(MEs, design , use = “p”);
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples)
# Will display correlations and their p-values
textMatrix = paste(signif(moduleTraitCor, 2), “\n(“,
signif(moduleTraitPvalue, 1), “)”, sep = “”);
dim(textMatrix) = dim(moduleTraitCor)
png(“step5-Module-trait-relationships.png”,width = 800,height = 1200,res = 120)
par(mar = c(6, 8.5, 3, 3));
# Display the correlation values within a heatmap plot
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = colnames(design),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste(“Module-trait relationships”))
dev.off()
#计算MM值和GS值并绘图
##切割,从第三个字符开始保存
modNames = substring(names(MEs), 3)
geneModuleMembership = as.data.frame(cor(dataExpr1, MEs, use = “p”));
## 算出每个模块跟基因的皮尔森相关系数矩
## MEs是每个模块在每个样本里面皿
## dataExpr1是每个基因在每个样本的表达量
MMPvalue = as.data.frame(
corPvalueStudent(as.matrix(geneModuleMembership), nSamples) ##计算MM值对应的P值
);
names(geneModuleMembership) = paste(“MM”, modNames, sep=””); ##给MM对象统一赋名
names(MMPvalue) = paste(“p.MM”, modNames, sep=””); ##给MMPvalue对象统一赋名
##计算基因与每个性状的显著性(相关性)及pvalue值
geneTraitSignificance = as.data.frame(cor(dataExpr1, design, use = “p”));
GSPvalue = as.data.frame(
corPvalueStudent(as.matrix(geneTraitSignificance), nSamples) ##计算GS值对应的pvalue值
);
names(geneTraitSignificance) = paste(“GS.”, colnames(design), sep=””);
names(GSPvalue) = paste(“p.GS.”, colnames(design), sep=””);
module = “brown”
column = match(module, modNames); ##在所有模块中匹配选择的模块,返回所在的位置
moduleGenes = moduleColors==module; ##==匹配,匹配上的返回True,否则返回false
png(“step6-Module_membership-gene_significance.png”,width = 800,height = 600)
par(mfrow = c(1,1)); ##设定输出图片行列数量,(1_1)代表行一个,列一丿
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]), ## 绘制MM和GS散点囿
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste(“Module Membership in”, module, “module”),
ylab = “Gene significance for Basal”,
main = paste(“Module membership vs. gene significance\n”),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)
dev.off()
#绘制TOM热图+模块性状组合图
geneTree = net$dendrograms[[1]]; ##提取基因聚类
dissTOM = 1-TOMsimilarityFromExpr(dataExpr1, power = 7); ##计算TOM距离
plotTOM = dissTOM^7; ##通过7次方处理进行标准化,降低基因间的误差
diag(plotTOM) = NA; ##替换斜对角矩阵中的内容为NA
TOMplot(plotTOM, geneTree, moduleColors, main = “Network heatmap plot, all genes”)
nSelect = 400 ##挑选需要提取的基因
set.seed(10); ##设置随机种子数,保证选取的随机性
select = sample(nGenes, size = nSelect); ##仿5000个基因中随机叿400丿
selectTOM = dissTOM[select, select]; ##提取挑出来的基因的TOM距离
# There’s no simple way of restricting a clustering tree to a subset of genes, so we must re-cluster.
selectTree = hclust(as.dist(selectTOM), method = “average”) ##对挑出来对基因TOM距离重新聚类
selectColors = moduleColors[select];
# Open a graphical window
sizeGrWindow(9,9)
# Taking the dissimilarity to a power, say 10, makes the plot more informative by effectively changing
# the color palette; setting the diagonal to NA also improves the clarity of the plot
plotDiss = selectTOM^7; ##对挑选出对基因TOM距离7次方处理
diag(plotDiss) = NA; ##替换斜对角矩阵中的内容为NA
png(“step7-Network-heatmap.png”,width = 800,height = 600)
TOMplot(plotDiss, selectTree, selectColors, ##绘制TOM图
main = “Network heatmap plot, selected genes”)
dev.off()
# 模块性状组合图
RA = as.data.frame(design[,2]); ##提取特定性状
names(RA) = “RA”
# Add the weight to existing module eigengenes
MET = orderMEs(cbind(MEs, RA))
# Plot the relationships among the eigengenes and the trait
sizeGrWindow(5,7.5);
par(cex = 0.9)
png(“step7-Eigengene-dendrogram.png”,width = 800,height = 600)
plotEigengeneNetworks(MET, “”, ##绘制模块聚类图和热图
marDendro =c(0,5,1,5), ##树类图区间大小谁宿
marHeatmap = c(5,6,1,2), cex.lab = 0.8, ##树类图区间大小谁宿
xLabelsAngle = 90) ##轴鲜艿90庿
dev.off()
#导出单个模块的网络结果
TOM = TOMsimilarityFromExpr(dataExpr1, power = 7); ##需要较长时间
# 选择brown模块
module = “brown”;
# Select module probes
probes = colnames(dataExpr1)
inModule = (moduleColors==module);
## 提取指定模块的基因名
modProbes = probes[inModule];
modTOM = TOM[inModule, inModule];
dimnames(modTOM) = list(modProbes, modProbes)
## 模块对应的基因关系矩
cyt = exportNetworkToCytoscape( ##导出单个模块的Cytoscape输入文件
modTOM,
edgeFile = paste(“CytoscapeInput-edges-“, paste(module, collapse=”-“), “.txt”, sep=””), ##输出边文件名
nodeFile = paste(“CytoscapeInput-nodes-“, paste(module, collapse=”-“), “.txt”, sep=””), ##输出点文件名
weighted = TRUE,
threshold = 0.02,
nodeNames = modProbes,
nodeAttr = moduleColors[inModule]
)
CytoscapeInput-edges-brown.txt,边文件
CytoscapeInput-nodes-brown.txt,点文件
最终小果成功完成了WGCNA分析,基本达到了复现,今天小果的分享就到这里啦!如果小伙伴有其他数据分析需求,可以尝试本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:http://www.biocloudservice.com/home.html,包括了WGCNA分析(http://www.biocloudservice.com/336/336.php),GEO数据下载(http://www.biocloudservice.com/371/371.php)等小工具,欢迎大家和小果一起讨论学习哈!!!!