MEGA是多序列比对的软件,并且可以进行绘制系统发育树。自1993年发布以来,MEGA共更新9个版本 (没有第八、九版),今年发布的MEGA 11为处理更大的数据集进行了优化。
软件的下载:
根据使用电脑系统的情况,可支持Win、Mac和Linux系统。下载链接如下:https://www.megasoftware.net/
 下载好的软件图标
下载好的软件图标
基本操作
数据导入
下载安装好 MEGA 11 后,首先打开软件。
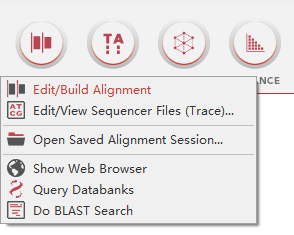
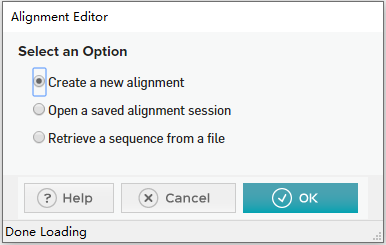
在 MEGA 11首页选择”ALIGN”,点击 “Edit/Build Alignment” ,会弹出一个对话框,选择”Create a new alignment”,根据需要比对的序列 (氨基酸序列或核苷酸序列),选择”DNA”或”Protein”。(序列可以在NCBI下载一个)
点击”Edit”,选择”Insert Sequence From File”导入我们需要比对的序列,序列文件格式为.fasta格式的哦~

序列比对
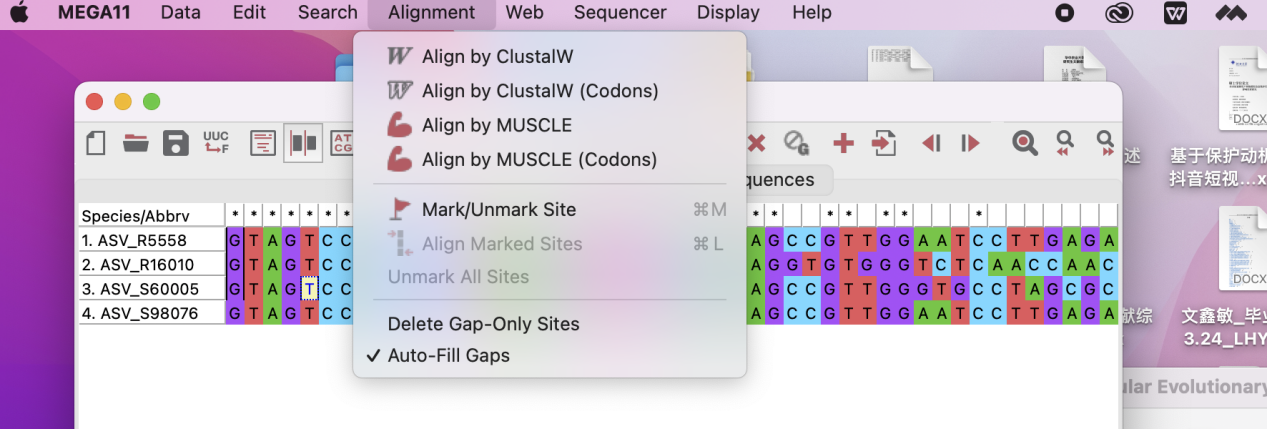
对目标序列进行多序列比较,可以使用ClustalW和MUSCLE,这里选择根据个人喜好和查阅相关的文件确定用什么算法进行比对。
比较结果如图所示:
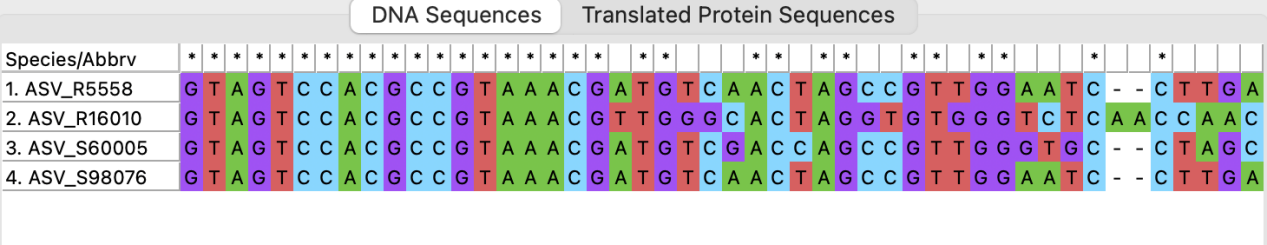 最后点击”Data”选择”Save Session”,保存序列比对的结果。
最后点击”Data”选择”Save Session”,保存序列比对的结果。
系统发育树
MEGA可以基于序列比对的结果进行进一步绘制发育树。点击”Data”,选择”Phylogenetic Analysis”进行系统发育分析。
然后,返回主页面,点击”PHYLOGENY”,构建系统发育树主要有三种方法,分别是最大似然法 (Maximum Likelihood)、邻接法 (Neighbor-Joining) 和最小进化法 (Minimum Evolution)。
这里补充一下有关的算法的介绍:
在系统进化学中,常用的树构建方法包括最大简约法(Maximum Parsimony, MP)、最大似然法(Maximum Likelihood, ML)、贝叶斯方法(Bayesian Inference, BI)和邻接法(Neighbor Joining, NJ)等。这些方法都有各自的优点和局限性,下面简单比较一下它们的特点:
最大简约法(MP):MP是一种基于字符分析的树构建方法,它假设在进化过程中尽量减少进化事件的数量。因此,MP方法的优点是计算速度快,适用于小样本和小数据集。缺点是对数据的缺失和多态性较敏感,而且在复杂模型中的应用效果不如其他方法。
最大似然法(ML):ML是一种基于模型的树构建方法,它利用概率模型计算数据的似然度,寻找最有可能产生观测数据的树。ML方法的优点是对数据的缺失和多态性较为鲁棒,而且在复杂模型中有较好的表现。缺点是计算复杂度高,需要较长的运行时间,且对于大数据集和复杂模型来说,可能会出现过拟合的情况。
贝叶斯方法(BI):BI是一种基于概率的树构建方法,它使用贝叶斯统计学原理估计树的后验概率。BI方法的优点是可以有效处理多态性和缺失数据,并且可以提供树的置信度。缺点是计算复杂度较高,需要长时间的运行,同时也需要选择合适的先验概率分布。
邻接法(NJ):NJ是一种基于距离的树构建方法,它利用序列之间的距离计算树的拓扑结构。NJ方法的优点是计算速度快、稳定性高、适用于大样本和大数据集。缺点是对于距离矩阵的错误或偏差比较敏感,因此需要合理选择距离计算方法。
需要注意的是,不同的树构建方法可能会产生不同的树结构和分支长度,因此选择合适的方法需要根据实际情况来确定。同时,多种方法结合使用或对比分析可以更好地提高分析结果的准确性。
一般我们在MEGA中选择第二个邻接法(Neighbor Joining, NJ)来绘制系统发育树,设置参数设置,Bootstrap method,1000次。
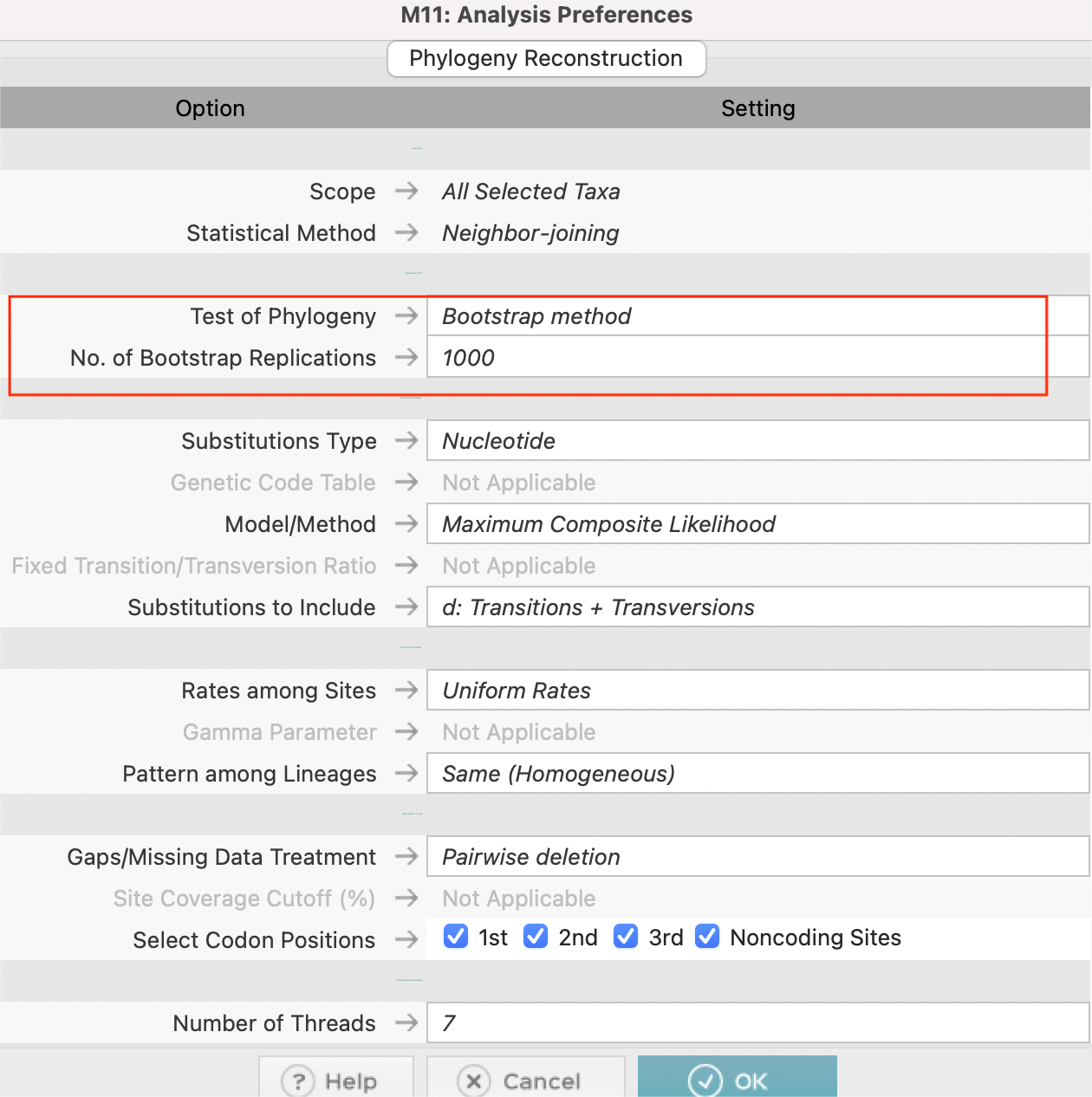
等待完成后,可以在这个页面进行修改一些参数,
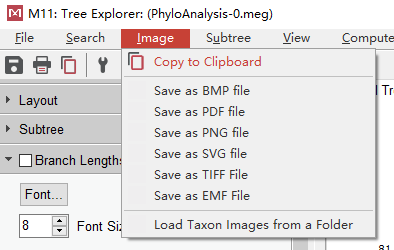
保存
点击”Copy to Clipboard”可保存为多种图片格式 (PDF、PNG、TIFF等),还可以选择”Copy to Clipboad”,直接复制到word中进行图片编辑(wps不可以)。
这样我们就学会了用MEGA这个软件啦,小伙伴们如果有什么问题就和小果讨论吧。