不知道小伙伴们是否理解Peak的含义呢? 这里小果就带大家理解一下ATAC-seq中的peak
首先先介绍一下ATAC-seq:ATAC-seq是一种测量染色质可及性的技术,利用转座酶Tn5在开放染色质区域切割DNA并插入测序接头,从而可以通过高通量测序检测到哪些区域是染色质开放的。那么ATAC-seq中的peak是指在某一位置或区域,测序信号达到最大值的现象,通常反映了某种基因调控元件(如启动子、增强子等)或转录因子结合位点的存在。ATAC-seq的peak分析可以帮助我们发现不同细胞类型或状态下,染色质可及性的变化,以及与之相关的转录调控机制。
例如下图:

图中的一个个突起的小山峰就是我们今天的主角peak了。
那peak怎么得到呢?又为什么要去注释呢?
小伙伴们是不是有点晕呢?哈哈哈不要着急,且听小果细细道来
首先我们的ATAC-seq的测序数据row data经过清理之后得到clean data,然后对clean data比对到基因组之后得到bam文件,经过排序,标记单端比对(双端测序),去除PCR重复等过程就会得到最终的final bam文件,然后就可以采用MACS2 软件来得到peak啦
那么为什么要对peak进行注释呢?Peak的注释是将peak与基因组上的已知元件(如基因、转录起始位点、启动子、增强子等)进行比较和对应,从而推断peak的功能和作用对象123。Peak的注释可以帮助我们理解peak在基因调控网络中的角色,发现与peak相关的基因和转录因子,以及探索不同细胞类型或状态下,peak的差异和变化。换句话说是因为单纯的peak只是一些冰冷的数据,只有通过注释,才能将peak匹配到一些基因元件上。
下面小果讲一下怎么用HOMER软件去注释peak
HOMER软件是一种用于分析和注释基因组上的peak的工具,主要用于ChIP-seq,ATAC-seq数据的处理。HOMER软件可以提供以下功能:
- 寻找和注释与转录因子或组蛋白修饰相关的peak
- 比较不同样本或条件下的peak差异
- 识别和分析转录因子结合位点的motif
- 评估peak在基因表达调控中的作用
- 可视化peak在基因组上的分布和特征
首先软件的安装:我们果断选择conda:
conda install -c bioconda homer
安装完HOMER之后使用configureHomer.pl完成HOMER软件的配置
- # 下载配置文件
- wget http://homer.salk.edu/homer/configureHomer.pl
- # 使用配置文件进行软件配置
- perl configureHomer.pl -install
如果是人类和小鼠等的数据就直接可以用configureHomer.pl来下载相应的参考基因组
以人的参考基因组为例:
- perl configureHomer.pl -install hg19
如果是自定义HOMER数据的话需要参考基因组的fasta文件和gtf文件,使用loadGenome.pl命令来自定义HOMER(需要HOMER版本4.4以上)
- loadGenome.pl -gtf test.gtf -name test -fasta test_assembly.fa -org null
接下来就是peak的注释啦,peak的注释需要用到annotatePeaks.pl 命令,输入文件可以是MACS2软件callpeak的bed文件(可以直接使用)或者是HOMER软件指定的peak文件格式。peak文件格式:使用Tab分隔,共五列,分别是 peak ID , chr , start , end ,strand
annotatePeaks.pl <Homer Peak/Positions file> <genome> 1> peak.anno.xls 2> anno.log
小果的代码如下:
annotatePeaks.pl test.bed Bomo 1> peak.anno.xlsx 2> anno.log
结果会生成一个xlsx文件,里面是注释的结果信息
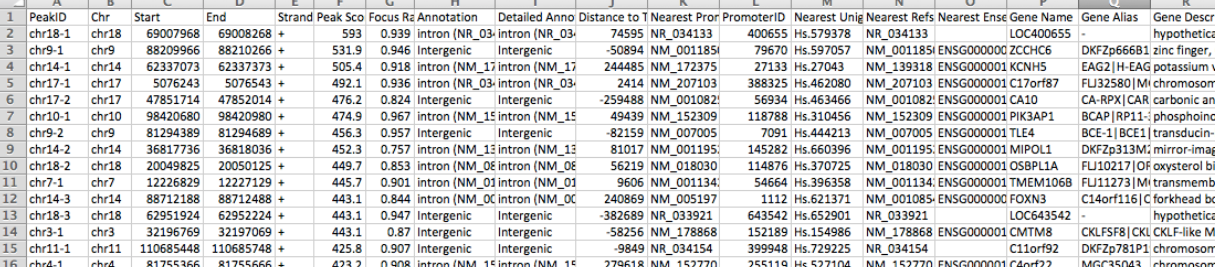
今天HOMER注释peak的学习就到这里啦,感兴趣的小伙伴可以找小果讨论哦,我们明天见咯~