bioawk简介
生信人一定用过三剑客awk、grep、sed,生信牛人——李恒,根据awk的源代码,进行修修补补专门用于生信的小工具。
作者:做生信的应该没有人不知道李恒大神了,鼎鼎大名的BWA在2009年到2019年短短10年的引用次数已经接近20K了,这样的引用次数对于生物软件来说绝对是数一数二的了。李恒的文章随随便便就是几千的引用。最高的两篇引用上万次的,分别是BWA和SAMtools对应的文章。真的很感谢大佬给予我们的便利,快来学一下这个软件的简便功能叭~
安装bioawk
首先,比bioawk是在Linux环境中使用的,推荐使用conda安装。一行命令就搞定,conda的使用要用-p指定安装目录。
安装命令: connda install -p ~/conda path/ bioawk -y
格式介绍
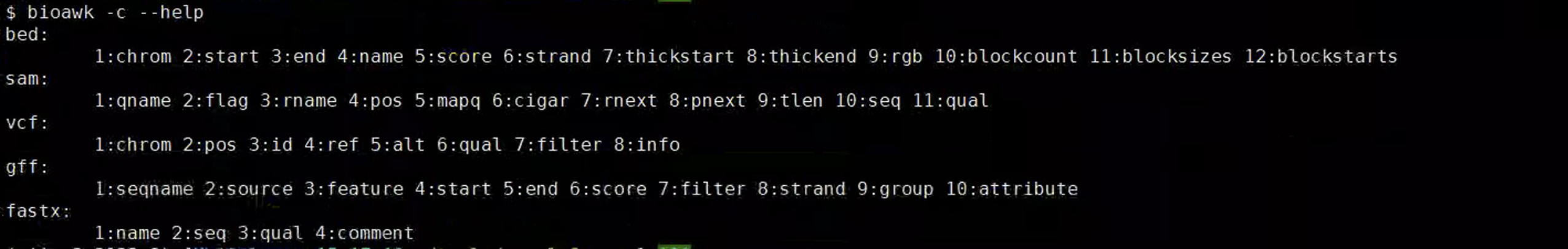
-c指定文件格式:常用的有bed、sam、vcf、gff、fastax
-c –help,查看可以用的格式,在使用过程中如果对格式不清楚,可以反复查看确定。
$ bioawk -c –help
bed:
1:chrom 2:start 3:end 4:name 5:score 6:strand 7:thickstart 8:thickend 9:rgb 10:blockcount 11:blocksizes 12:blockstarts
sam:
1:qname 2:flag 3:rname 4:pos 5:mapq 6:cigar 7:rnext 8:pnext 9:tlen 10:seq 11:qual
vcf:
1:chrom 2:pos 3:id 4:ref 5:alt 6:qual 7:filter 8:info
gff:
1:seqname 2:source 3:feature 4:start 5:end 6:score 7:filter 8:strand 9:group 10:attribute
fastx:
1:name 2:seq 3:qual 4:comment
安装好后,就可以着手使用了,小果总结了一些在生信分析过程中可以应用的
案例
下面就通过一个例子来展示
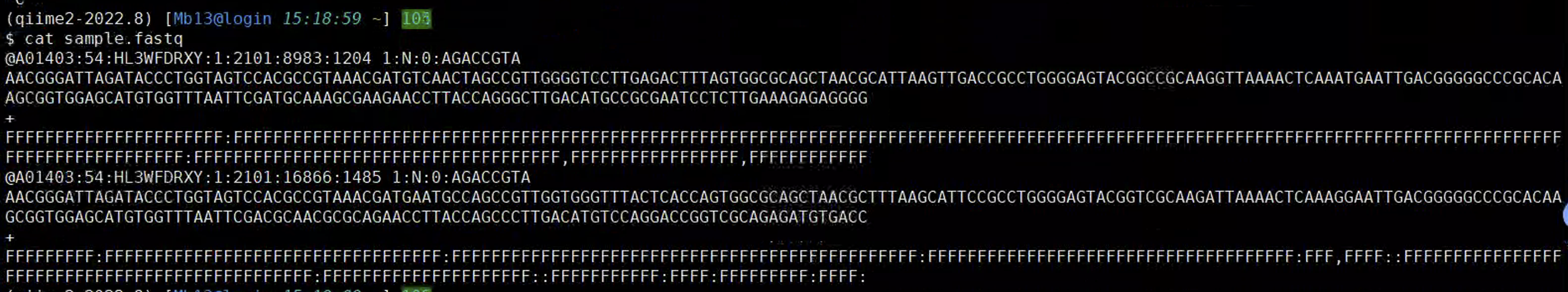
#首先准备一个fastq格式的文件
$ cat sample.fastq
@A01403:54:HL3WFDRXY:1:2101:8983:1204 1:N:0:AGACCGTA
AACGGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGTCAACTAGCCGTTGGGGTCCTTGAGACTTTAGTGGCGCAGCTAACGCATTAAGTTGACCGCCTGGGGAGTACGGCCGCAAGGTTAAAACTCAAATGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTTTAATTCGATGCAAAGCGAAGAACCTTACCAGGGCTTGACATGCCGCGAATCCTCTTGAAAGAGAGGGG
+
FFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF,FFFFFFFFFFFFFFFFF,FFFFFFFFFFFF
@A01403:54:HL3WFDRXY:1:2101:16866:1485 1:N:0:AGACCGTA
AACGGGATTAGATACCCTGGTAGTCCACGCCGTAAACGATGAATGCCAGCCGTTGGTGGGTTTACTCACCAGTGGCGCAGCTAACGCTTTAAGCATTCCGCCTGGGGAGTACGGTCGCAAGATTAAAACTCAAAGGAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGTTTAATTCGACGCAACGCGCAGAACCTTACCAGCCCTTGACATGTCCAGGACCGGTCGCAGAGATGTGACC
+
FFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFF,FFFF::FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFF::FFFFFFFFFFF:FFFF:FFFFFFFFF:FFFF:
统计序列的长度
#-c fastx 指示出相应的格式,打印出name列和seq的长度。
$ bioawk -c fastx ‘{print $name”\t” length($seq)}’ sample.fastq

是不是很方便,接下来再来探索以下其它的功能叭~
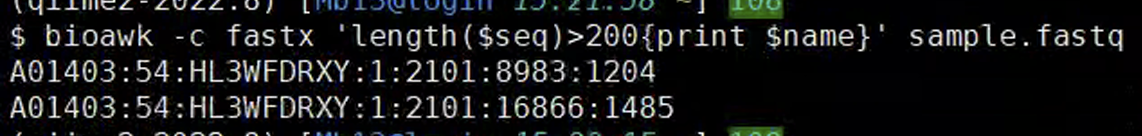
取特定长度bp的序列
取出大于200bp序列的id ,序列等等
$ bioawk -c fastx ‘length($seq)>200{print $name}’ sample.fastq
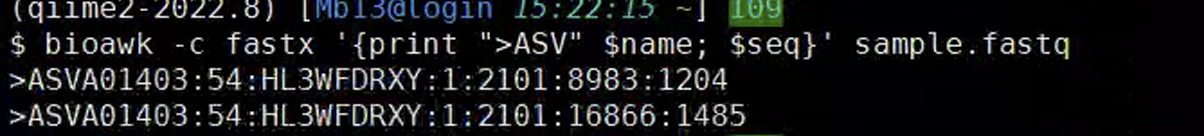
对序列添加标签
$ bioawk -c fastx ‘{print “>ASV” $name; $seq}’ sample.fastq
统计GC含量
$ bioawk -c fastx ‘{print $name,gc($seq)}’ sample.fastq
fastq转fasta
按bioawk的原理,这个转化将变得非常的简单,小伙伴们可以自己思考一下,或者可以用你熟悉的编程语言实现也是可以的哦~
$ bioawk -c fastx ‘{print “>”$name;print $seq }’ sample.fastq

是不是很简单的一句,真的是太方便了,再接触这个软件之前小果也尝试用其它的方式进行fastq与fasta之间的转化,例如;
cat sample.fastq |paste – – – – |cut -f 1,2|sed ‘s/^@/>/’|tr “\t” “\n”|awk ‘{print $1}’>gene.fa

根据序列id提取序列
bioawk -cfastx ‘BEGIN{while((getline k <“id.txt”)>0)i[k]=1}{if(i[$name])print “>”$name”\n”$seq}’ sample.fastq

这里的id.txt存放的是不带“>”的id
过滤掉短于10bp的reads
bioawk -c fastx ‘length($seq) > 10 {print “@”$name”\n”$seq”\n+\n”$qual}’ input.fastq
处理SAM文件
将未比对上的序列提取出来:bioawk -c sam ‘and($flag,4)’ input.sam
将比对上的(mapped)序列提取出来:bioawk -c sam -H ‘!and($flag,4)’ input.sam
根据sam文件创建fasta:bioawk -c sam ‘{ s=$seq; if(and($flag,16)){s=revcomp($seq)} print “>”$qname“\n”s’ input.sam > out.fasta
#这里sam可能会存在反向序列,如果flag=16为反向序列,需要先用revcomp进行反转后再赋值给s,最后打印出名字和相应的序列即可。
总结
根据上述的实践操作,可以直观的感受到小小的软件,但涵盖的功能是非常的强大的。在数据处理中不免会遇到各种各样的问题,有时候写代码会很繁琐并且容易出现bug。不妨在平时多多积累一些好用的生信软件协助我们解决问题。这种大佬开发的专门用于生物信息学分析的软件,不仅能够快速的了解生物信息学中的文件格式,并且上手简单,非常适合小白的学习。
好了,这样我们小软件的分享就到这里叭。小伙伴们如果有什么问题就和小果讨论吧。