大家好,小果又来啦!这次带大家过一下Hi-C的数据分析流程
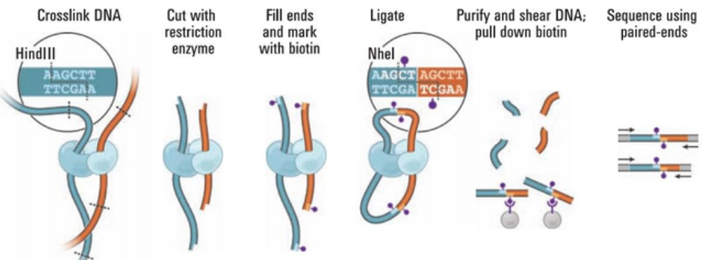
上图是Hi-C测序的原理图,大概可以分为一下几步:
- 利用甲醛固定细胞,使得蛋白质和DNA固定,蛋白与蛋白之间进行交联
- 进行酶切(HindIII),使得交联两侧产生粘性末端
- 末端修复,引入生物素标记,使用DNA连接酶连接临近的DNA片段
- 蛋白酶消化解除交联,纯化DNA并随机打断,亲和素磁珠捕获带有生物素标记的片段,进行建库
- 进行高通量测序
原理懂了,小果就带大家开始走流程咯
首先是下载测试数据集(参考基因组和Hi-C数据),代码如下:
wget https://s3.amazonaws.com/4dn-dcic-public/hic-data-analysis-bootcamp/input_R1.fastq.gz
wget https://s3.amazonaws.com/4dn-dcic-public/hic-data-analysis-bootcamp/input_R2.fastq.gz
gunzip input_R1.fastq.gz
gunzip input_R2.fastq.gz
# bwa genome index
wget https://s3.amazonaws.com/4dn-dcic-public/hic-data-analysis-bootcamp/hg38.bwaIndex.tgz
tar -xzf hg38.bwaIndex.tgz
rm hg38.bwaIndex.tgz
# chromsizes
wget https://s3.amazonaws.com/4dn-dcic-public/hic-data-analysis-bootcamp/hg38.mainonly.chrom.size
经过上面的下载,可以得到两个fastq文件和参考基因组文件还有参考基因组的chrom size 文件
下面就开始第一步:mapping
使用bwa软件比对接着使用samtools将比对后的sam文件转换为bam文件,小果的代码如下:
bwa mem -SP5M -t 32 hg38.fasta input_R1.fastq input_R2.fastq | samtools view -bhS – > output.bam
下面是参数的解释:
-t : 进程数
-SP5M :Hi-C specific options
比对之后可以使用samtools view 结合less命令来观察生成的bam文件:
samtools view out.bam | less -S
接着是第二步过滤,在实际的比对中,可能只有一端能比对到基因组上,另一端无法识别到基因组上,这种情况归为Singleton,如果两侧数据都能够唯一比对到基因组上,则归类为Unique mapped reads ,对于动物基因组,unique mapped reads占测序量50%以上可以接受。对于植物样本,尤其是重复序列较多的样本,Unique mapped reads比例可能急剧降低。在获取Unique mapped reads 之后,要进行进一步过滤来筛选vaild pairs。
根据Hi-C的原理,仅当两个read来源于不同的酶切片段时才被认为是标准的文库片段。即两个read分别匹配到不同的酶切片段上,且实际片段大小符合理论的片段大小,这样才会将该reads归类到vaild pairs中
过滤步骤使用的软件pairtools,主要是生成.pairs文件(具体.pairs格式可以看这个链接:https://github.com/4dn-dcic/pairix/blob/master/pairs_format_specification.md)。小果的代码如下
- samtools view -h output.bam | pairtools parse -c hg38.mainonly.chrom.size -o parsed.pairsam
- pairtools sort –nproc 8 -o sorted.parsed.pairsam parsed.pairsam
- less -S sorted.parsed.pairsam
下面的步骤是去重,去重使用到的软件仍然是pairtools,小果的代码如下:
pairtools dedup –mark-dups -o deduped.sorted.parsed.pairsam sorted.parsed.pairsam
接着下一个步骤是select,即根据pairs的类型筛选pairs,具体的pairs类型可以去看pairtools的官网嗷
小果的代码如下:
pairtools select ‘(pair_type == “UU”) or (pair_type == “UR”) or (pair_type ==”RU”) ‘ -o filtered.deduped.sorted.parsed.pairsam deduped.sorted.parsed.pairsam
上述代码筛选出UU,UR和RU类型的pairs
下一个步骤是拆分文件:将最终得到的.pairsam文件拆成.pairs文件和.sam文件,使用到的软件还是老朋友pairtools!
小果的代码是这样的:
pairtools split –output-pairs output.pairs filtered.deduped.sorted.parsed.pairsam
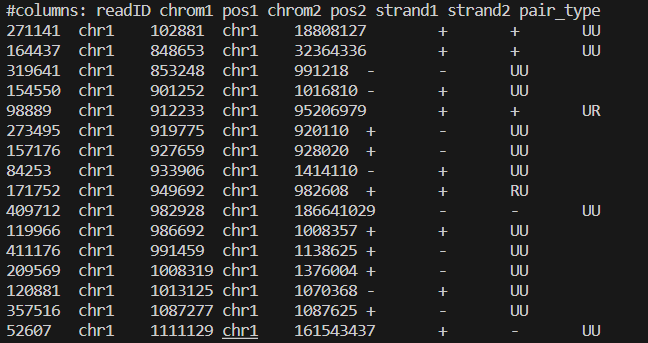
less -S output.pairs
这是小果的output.pairs文件的样子
快结束啦,大家不要着急嗷
下面就是创建索引啦:对pairs文件创建索引,这个步骤用到的软件是pairix
下面是小果的代码:
- 对.pairs文件进行压缩,得到gz文件
bgzip output.pairs
- 使用pairix建立索引
pairix -f output.pairs.gz
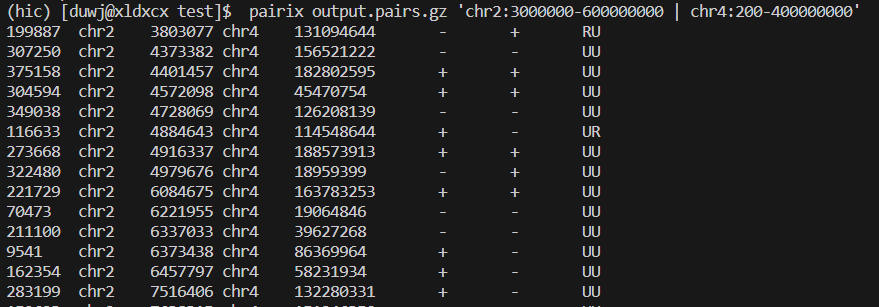
- 查询染色体某区域的的pairs
pairs pairix output.pairs.gz ‘chr2:3000000-600000000 | chr4:200-400000000’
下面是第三步的运行结果
下一步就是生成contact矩阵,要用到的软件是cooler,大家使用conda安装即可。输入文件是.pairs文件和chromesize文件,输出则是cool文件
小果的代码是这样滴:
cooler cload pairix hg38.mainonly.chrom.size:50000 output.pairs.gz output.cool
其中chromesize文件后的:50000 是指bin(互作基本单位)为50000
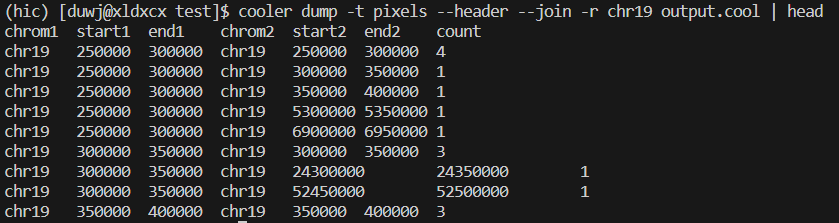
可以使用下面命令查看.cool文件(contact矩阵)
cooler dump -t pixels –header –join -r chr19 output.cool
小果生成的contact矩阵如下图所示:
今天的关于Hi-C分析流程的学习就到这里啦,感兴趣的小伙伴可以找小果讨论,有感觉生信分析复杂的小伙伴可以直接使用我们的生信小工具哦,链接在这:http://www.biocloudservice.com/home.html,我们明天见咯~