今天小果在下载一个GEO数据集的时候发现该芯片平台注释信息不全,没有相关的基因注释信息只有探针信息,没有基因注释信息就无法提取基因表达矩阵,小果将通过重新注释的方法来获得基因表达矩阵,有需要的小伙伴,可以跟着小果一起开始今天的学习。
- 如何进行芯片数据重注释?
小果在进行GSE92681数据集下载的时候发现该数据集没有基因注释信息,只包含探针ID,SEQUENCE和transcript类型,如下图:

遇到这种情况大多数小伙伴是不是没有办法啦!不用着急,小果来为小伙伴解决这个问题,一般通过芯片重新注释来解决这个问题,一般通过这几个步骤来进行重新注释,第一步,需要准备芯片上的探针序列和转录本DNA序列,探针序列已经存在,需要下载转录本DNA序列,下载于GENCODE(https://www.gencodegenes.org/human/),

小果下载最新版本的转录本DNA序列(gencode.v43.transcripts.fa),转录本id包括了Gene symbol和biotype。
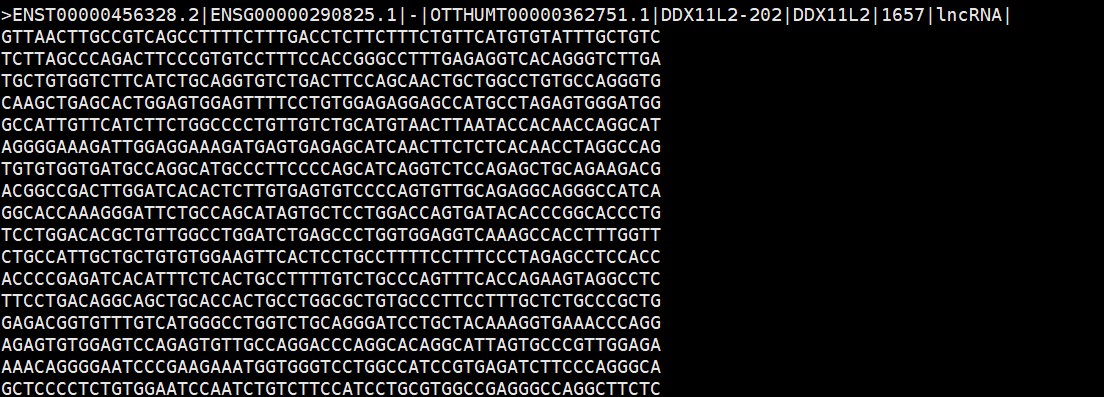
第二步,准备好了探针序列和转录本DNA序列后,可以选择blast或者seqman软件进行序列比对,本推文小果利用seqman软件进行比对,seqman相比blast软件,速度要很快,使用参数比较简单,最后根据序列比对结果提取探针id,Gene symbol和biotype对应关系文件。
#安装seqman,网址:http://www-personal.umich.edu/~jianghui/seqmap/

#下载seqman安装包
wget https://jhui2014.github.io/seqmap/download/seqmap-1.0.13-src.zip
#解压seqmap压缩包
unzip seqmap-1.0.13-src.zip
#进行编译
cd seqmap-1.0.13-src
make
#添加环境变量
export PATH=/media/desk16/wangd/seqmap-1.0.13-src:$PATH
source .bashrc
通过以上步骤就可以成功的安装seqman软件啦!
第三步,合并表达矩阵和探针id,Gene symbol和biotype文件,获得最终的基因表达矩阵文件。
这就是小果为大家展示的芯片重注释步骤,通过三步就可以该分析,接下来小果将开始今天的实操。
- 准备需要的R包
#安装需要的R包
BiocManager::install(“GEOquery”)
install.packages(“tidyverse”)
#加载需要的R包
library(GEOquery)
library(tidyverse)
3.GSE92681数据集表达矩阵和注释信息下载
#GSE92681数据集下载
GSEdata <- getGEO(“GSE92681”, GSEMatrix=T, AnnotGPL=FALSE)
eset <- GSEdata[[1]]
#提取表达矩阵,通过exprs函数提取,行名为探针ID,列名为样本名
exprSet <- as.data.frame(exprs(eset))
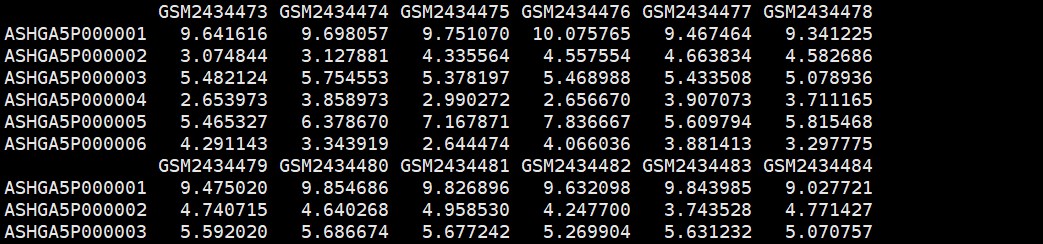
# 提取GSM ID作为样品名,pData函数来获取样本信息
GSMID <- pData(eset)[,c(1,2,8,10,12)]
#利用fData函数提取芯片注释信息,发现没有基因注释信息
annotation<-fData(eset)
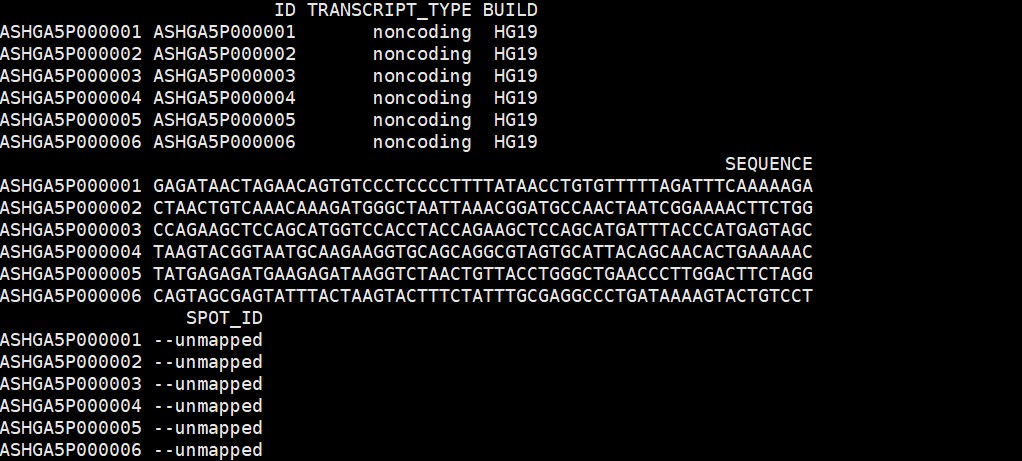
#只保留探针ID和序列
gpl <- annotation[,c(1,2,4)]
gp <- dplyr::filter(gpl,!is.na(SEQUENCE))
#获得探针对应的序列
gpl <- paste0(‘>’,gpl$ID,’\n’, gpl$SEQUENCE)
#保存全部序列,运行下面这行:
write.table(gpl,’GPL.fasta’, quote = F, row.names = F, col.names = F)
4.GSE92681数据集重注释
#利用seqmap软件将转录本的DNA序列文件和芯片上探针的序列进行序列比对
seqmap软件参数说明:
0代表匹配探针错误个数0
GPL.fasta 需要比对的探针序列
gencode.v43.transcripts.fa 转录本的fasta文件
seqmap_gene.txt 输出文件,比对结果文件
system(‘seqmap 0 GPL.fasta gencode.v43.transcripts.fa seqmap_gene.tmp /output_all_matches’)
# seqmap_gene.tmp,生成的比对结果文件,需要关注第一列转录本序列id和第四列探针id
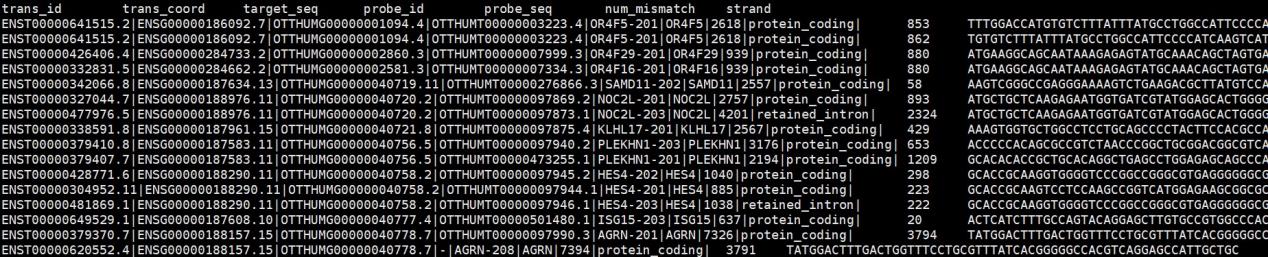
gplTOgene <- read_delim(‘seqmap_gene.tmp’,delim = ‘\t’)
#提取第1列和第4列数据
gplTOgene <- gplTOgene[,c(1,4)]
#将第一列数据分割获得gene gsym
gplTOgene$gsym<-unlist(lapply(gplTOgene$trans_id,function(x) strsplit(as.character(x),”\\|”)[[1]][6]))
#将第一列数据分割获得biotype
gplTOgene$biotype<-unlist(lapply(gplTOgene$trans_id,function(x) strsplit(as.character(x),”\\|”)[[1]][8]))
gplTOgene$gsym_biotype <- paste(gplTOgene$gsym,gplTOgene$biotype,sep = ‘|’)
#输出三列,探针ID、gene symbol、biotype
gplTOgene <- gplTOgene[,c(2:4)]
#去除重复
gplTOgene <- distinct(gplTOgene)
write_csv(gplTOgene,’gplTOgene.csv’)
#提取mRNA芯片注释信息,也可以选择提取LcRNA注释信息
mRNA.probe1 <- filter(gplTOgene,biotype ==”protein_coding”)
#获得唯一的探针id
uni <- names(table(mRNA.probe1$probe_id)[table(mRNA.probe1$probe_id) == 1])
mRNA.probe2 <- mRNA.probe1[match(uni,mRNA.probe1$probe_id),]
#获得一个gene对应两个探针的Gene symbol
nProbes <- names(table(mRNA.probe2$gsym)[table(mRNA.probe2$gsym) >= 2])
mRNA.probe3 <- mRNA.probe2[mRNA.probe2$gsym %in% nProbes,]
mRNA.probe4 <- mRNA.probe3[order(mRNA.probe3$gsym),]
#保存芯片id,Gene symbol和biotype三者关系文件
write.table(mRNA.probe4,”mRNA.probes.txt”, quote = F,row.names = F, col.names = T)
mRNA.probe4 <- read.table(“mRNA.probes.txt”, header = T)
#将行名转化为列名probe_id
exprSet<-rownames_to_column(exprSet,var=”probe_id”)
#合并注释信息和表达矩阵文件
mRNA.probe.expr <- merge(mRNA.probe4[,1:2],exprSet , by = “probe_id”)
#将列名转化为行名
row.names(mRNA.probe.expr) <- mRNA.probe.expr$probe_id
#去除probe_id列
mRNA.probe.expr$probe_id <- NULL
# 计算多个探针对应同一个基因表达量的中位数值
mRNA.expr_uniq <- aggregate(.~gsym,mRNA.probe.expr, median)
# 将 gene symbol 列转化为行名
rownames(mRNA.expr_uniq) <- mRNA.expr_uniq$gsym
#去除gene symbol 列
mRNA.expr_uniq$gsym <- NULL
#获得重新注释的mRNA(protein_coding)表达矩阵文件
GSE92681<-mRNA.expr_uniq
#保存结果文件
write.table(GSE92681,file=”GSE92681_gene_expr.txt”,col.names=T,row.names=T,quote=F,sep=”\t”)
- 结果文件
GSE92681_gene_expr.txt
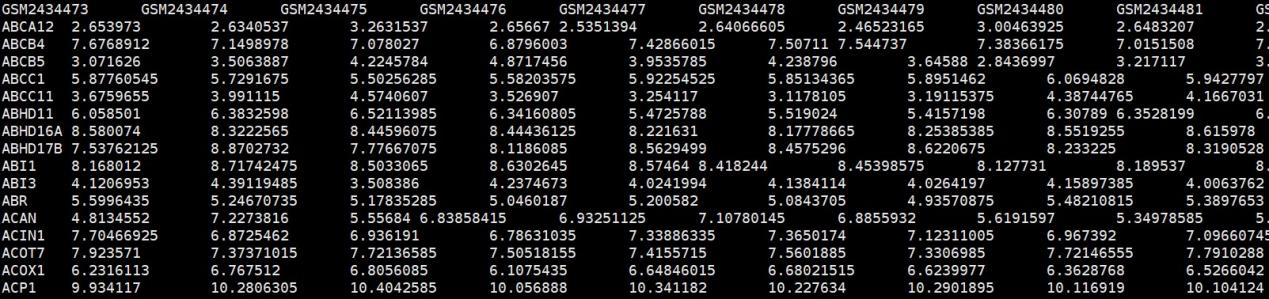
该结果文件进行重注释后获得的基因表达矩阵,行名为Gene symbol,列名为样本名。
今天小果的分享就到这里啦!如果小伙伴有其他数据分析需求,可以尝试本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:http://www.biocloudservice.com/home.html,包括了GEO数据下载(http://www.biocloudservice.com/371/371.php)等小工具,欢迎大家和小果一起讨论学习哈!!!!