小果最近研究三维基因组并且已经得到了contact矩阵(cool文件),那么怎么做出来美观的可视化图呢,像下面的图一样
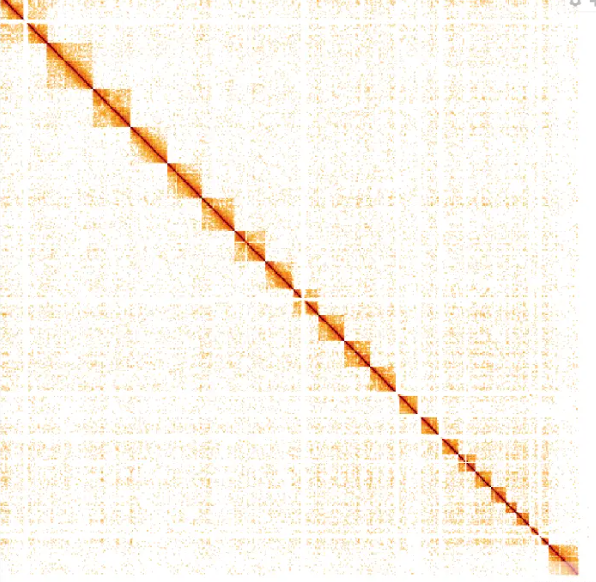
想学的小伙伴们就跟着小果一起来看吧~
首先向大家介绍一下jupyte notebook:jupyter是一个基于web的交互式计算平台。jupyter notebook是一个原始的web应用,用于创建和分享包含代码、方程、文本、可视化、交互式仪表盘和其他媒体的计算文档。jupyter支持超过40种编程语言,包括Python, R, Julia和Scala。你可以使用pip或conda等工具安装jupyter,或者在浏览器中在线尝试jupyter。
因为小果使用的是服务器,所以小果先在服务器上安装了一个jupyter notebook(如果大家是安装的anaconda的话就不用安装了哦,anaconda自带jupyter notebook),如果是miniconda的话则需安装,anaconda和miniconda的区别在下面咯
anaconda和miniconda的区别主要有两个方面:
- 包的数量:anaconda是一个包含了超过150个数据科学和机器学习包的完整的Python发行版,而miniconda只包含了conda、Python和一些其他的包。因此,anaconda的安装文件比较大,需要更多的磁盘空间,而miniconda的安装文件比较小,更加灵活。
- 界面的类型:anaconda提供了一个图形用户界面(GUI)叫做Navigator,可以通过它来创建环境、安装包、启动开发应用等。而miniconda只提供了一个命令行界面(CLI),需要通过输入命令来管理环境和包。
使用conda安装jupyter:
conda install jupyter
下面需要生成配置文件:
jupyter notebook –generate-config
然后会得到:
Writing default config to: /root/.jupyter/jupyter_notebook_config.py
说明了配置文件的地址,然后使用vim打开修改配置文件:
- c.NotebookApp.ip=’*’ #
- c.NotebookApp.ip=’0.0.0.0′
- c.NotebookApp.open_browser =False # 设置是否自动打开浏览器
- c.NotebookApp.port =8888 # 设置端口,可以自己更改
- c.NotebookApp.allow_remote_access = True
之后在命令行输入jupyter notebook可以启动jupyter notebook ,如果需要保持jupyter notebook挂起的话可以使用nohup : nohup jupyter notebook > jupyter.log 2>&1 &
如果需要结束jupyter进程的话可以使用一下代码:
ps -aux | grep jupyter | awk ‘{print $2}’ | xargs kill -9 -s
启动jupyter notebook之后,浏览器输入相应的ip和端口即可进入,首次进入需要输入token,token会在命令行中出现,输入即可
进入后发现只有python3一个kernel,如果想使用其他虚拟环境该怎么办呢
可以参考一下流程:
- 首先在你想要使用的虚拟环境中安装ipykernel包
conda activate YOUR_ENV_NAME
conda install ipykernel
- 在你安装jupyter notebook的环境里面运行以下代码:
python -m ipykernel install –user –name YOUR_ENV_NAME –display-name YOUR_ENV_NAME
这样再次打开就会出现你想使用的虚拟环境啦
上面是环境的配置,下面开始可视化cool文件
首先我们要使用higlass-python这个python包
使用pip安装:
pip install jupyter higlass-python
下面是关于higlass软件的一些介绍:higlass是一个用于可视化大规模数据的网页浏览器,最初设计用于处理海量的基因组数据集。higlass基于类似于基因组浏览器和现代在线地图(所谓的滑动地图)的框架,形成了一个快速、灵活和可扩展的多尺度数据浏览器。higlass支持多种1D和2D的轨迹类型,可以显示不同文件格式的数据。higlass使用D3.js、Pixi.js、React等开源组件进行数据可视化和交互。
下面就是可视化分析的代码啦
import higlass as hg
# 加载文件
tileset = hg.cooler(“software/HiC-Pro/Utils/test/.output.cool”)
track = tileset.track(“heatmap”)
view = hg.view(track, width=6)
# 服务器运行需要加,不然运行不出来
tileset = hg.remote(
uid=”CQMd6V_cRw6iCI_-Unl3PQ”,
server=”http://higlass.io/api/v1/”,
)
view = hg.view(
hg.track(“top-axis”),
tileset.track(“heatmap”, height=250).opts(
valueScaleMax=0.5,
),
)
view
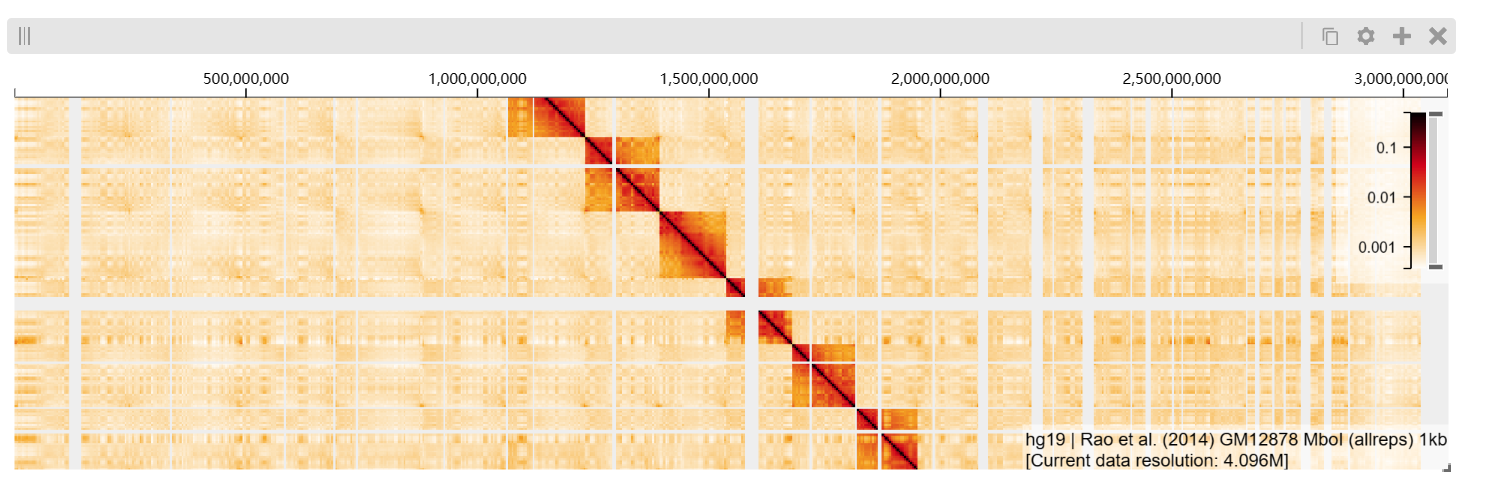
下面是小果的运行结果,大家一起试一试吧
今天的关于higlass可视化的学习就到这里啦,感兴趣的小伙伴可以找小果讨论,有感觉生信分析复杂的小伙伴可以直接使用我们的生信小工具哦,链接在这:http://www.biocloudservice.com/home.html,我们明天见咯~