全基因组关联研究(Genome-Wide Association Study,GWAS)迅速发展,为探索疾病的发病机制做出了突出贡献,也为精准医学的发展奠定基础。在进行GWAS分析时,经常会使用到PLINK软件,对于新手来说可能掌握起来比较困难,
PLINK是一个免费的开源全基因组关联分析工具集,旨在以高效率计算的方式执行一系列基本的、大规模的分析。PLINK的重点分析对象是基因型或者表型数据,可以为后续的可视化、注释和结果存储提供一些支持。
【1】工具下载:https://www.cog-genomics.org/plink2/
首先我们打开页面,确定好咱们Windows的操作系统(一般都是64位的,红色标记处),点击“download”后开始下载,如下图所示:
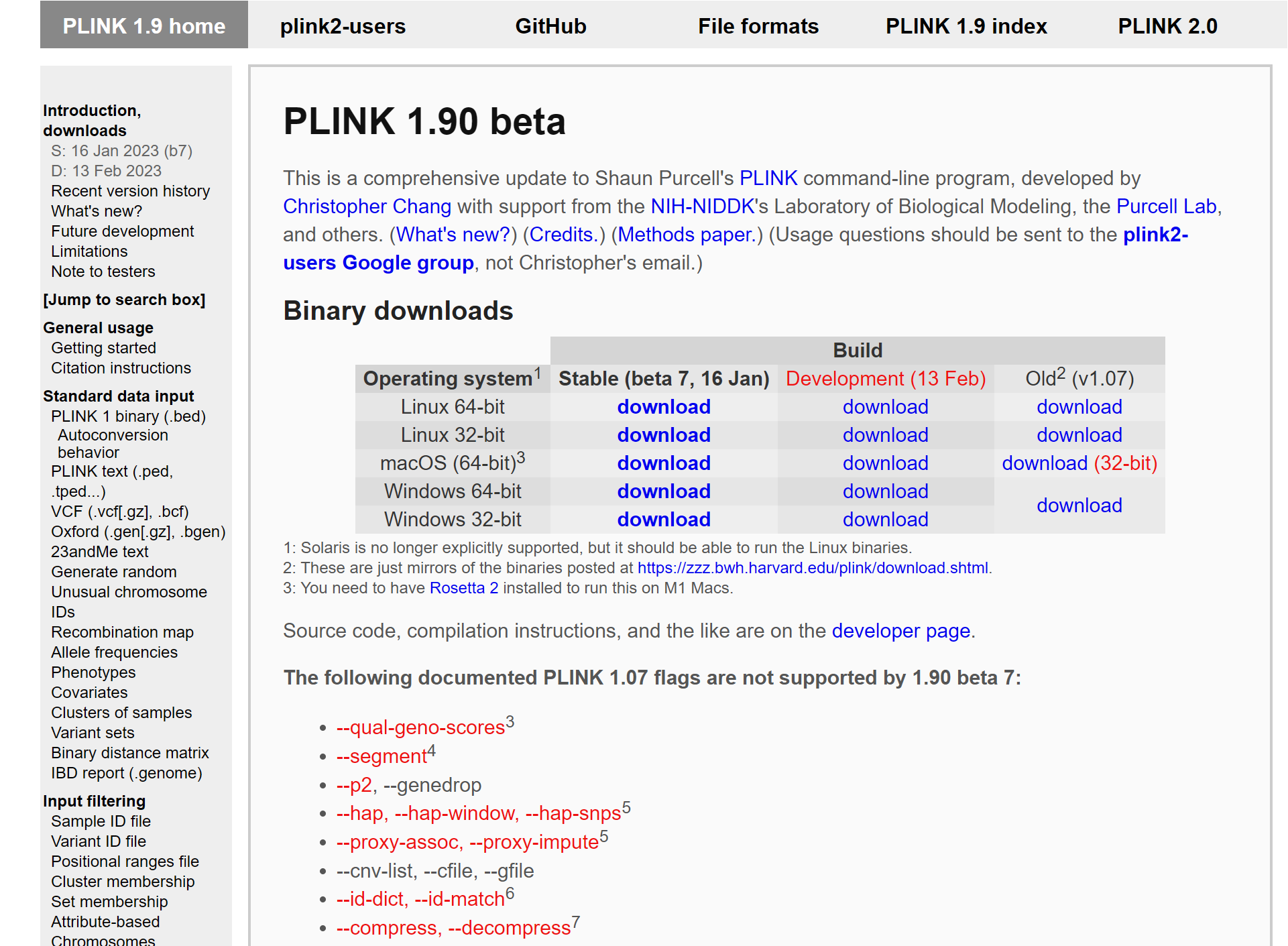
【2】使用手册:https://zzz.bwh.harvard.edu/plink/index.shtml

所以首先小果将和大家分享PLINK文件的基本格式。小果在这里讲到的PLINK文件主要有三类,即bed、bim和fam文件。其中bed是存储基因型信息的,bim文件则是存储每个遗传变异(通常是SNP)的相关信息,最后的fam存储的是样本信息,接下来小果将一一介绍。
输入文件准备
- ped
1.1 ped 包含样本的谱系信息和基因型信息
1.2 ped 必须与fam 文件一起,前6个字段与fam文件对应
1.3 使用recode 产生ped文件
举例:每一行为一个样本
 第一列 家系编号(Family ID)
第一列 家系编号(Family ID)
第二列 家系内部编号(Individual ID)
第三列 父系编号 (Paternal ID)
第四列 母系编号 (Maternal ID)
第五列 性别编号(Sex 【1=male; 2=female;other=unknown】)
第六列 表型值(Phenotype)
- map
举例: 文件中必须且仅包含四列
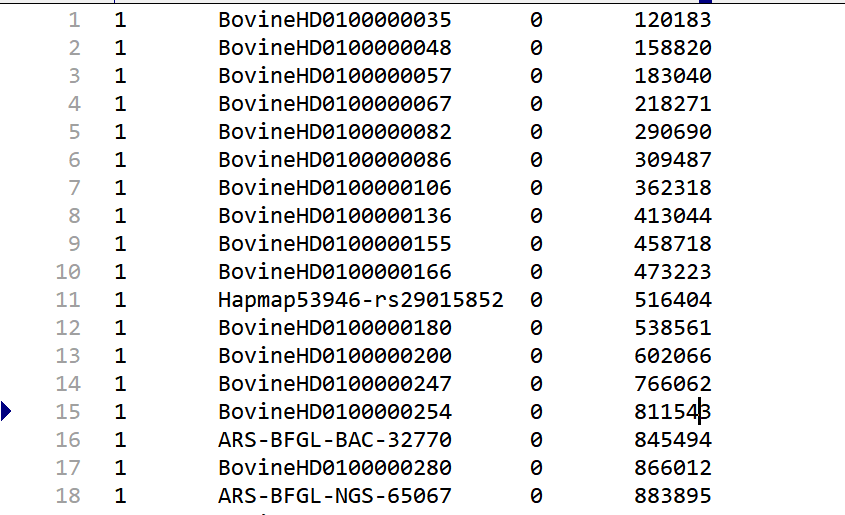
第一列代表染色体
第二列代表snp的名称
第三列代表摩尔距离,一般用不上
第四列代表物理距离,这个有用的
- bed
文件bed主要是存储等位基因信息,该格式的文件主要包含了基因型的双等位基因变异。必须附带.bim和.fam文件。用–bfile加载;可以使用–make-bed命令生成。注意的是该格式和我们平时接触含有基因组位置的bed格式文件不同。在基因型信息中,每一行是一个个体,每一列就是一个变异。其中0、1、2分别对应了aa、Aa或aA和AA。
- bim
bim文件是存储每个遗传变异(通常是SNP)的相关信息文件,是一个没有题头的文本文件,每一行代表一个遗传变异,共计有6列,其信息如下:
4.1 第一列是染色体信息
4.2 第二列是snp的名字
4.3 第三列是摩尔距离,可以用0
4.4 第四列是物理距离
4.5 第五列是次要等位基因
4.6 第六列是主要等位基因
- fam
关于fam文件,fam存储的是样本信息,它也是一个没有题头的文本文件,每一行代表一个样本,共计6列,其信息如下:
5.1 fam文件记录了每个样本家系的信息
5.2 fam文件也是必须和bed bim文件一起使用
5.3 主要有6 列
关于PLINK文件的基本格式就介绍完毕了,希望大家能牢记各个文件的结构信息,这在后续的数据分析中非常重要。
欢迎使用:云生信生物信息学平台( http://www.biocloudservice.com/home.html),了解更多生信知识与技巧分析。
请期待小果的下一期分享。