Hello大家好,小果又来啦
小果今天使用HiC-Pro给大家分析一下Hi-C的数据
首先熟悉三维基因组的小伙伴们肯定都知道Hi-C数据的处理软件里面HiC-Pro和juicer是比较流行的,那么这两个软件的区别和侧重点是什么呢?且听小果细细道来
- HiC-Pro是一个灵活和高效的管道,可以从原始测序数据到规范化的联系矩阵。它支持多种限制性酶切位点,可以并行处理多个样本,可以检测和过滤PCR重复,可以生成多种格式的输出文件。
- juicer由两部分组成:从原始数据到创建Hi-C文件的pipeline和后续分析工具。它可以处理terabase规模的Hi-C数据集,自动注释Loops和Domains,与多个集群操作系统和Amazon Web Services兼容。它还可以利用GPU进行峰值调用。
- 两者的主要区别在于juicer提供了一个一键式的系统,而HiC-Pro需要用户自己配置参数和运行步骤。juicer还提供了更多的后续分析工具,如3D-DNA,可以用来辅助基因组组装。HiC-Pro支持多种限制性酶切位点,而juicer只支持HindIII和MboI两种。HiC-Pro可以生成多种格式的输出文件,如bed, matrix, pairs等,而juicer只生成hic文件。HiC-Pro可以让用户自己配置参数和运行步骤,有更多的灵活性和可定制性,而juicer是一个一键式的系统,有一些默认的设置和限制。
可能有小伙伴对第二点中的terabase表示疑问,不知道是什么,其实terabase是一个计量单位,表示10的12次方个字节,也就是一万亿个字节。它通常用来表示大规模的数据存储或传输。例如,一个terabase的Hi-C数据集就是包含了一万亿个Hi-C测序对的数据集
听说HiC-Pro软件安装坑很多?小果手把手教大家安装HiC-Pro!
- 首先创建自己的软件目录然后进入,接着下载HiC-Pro:
wget https://github.com/nservant/HiC-Pro/archive/refs/tags/v3.1.0.tar.gz
- 解压出来:
tar -zxvf HiC-Pro-3.1.0.tar.gz
- 使用conda根据yml文件创建新的环境
conda env create -f YOUR_PATH/HiC-Pro-3.1.0/environment.yml -n hicpro
- 切换到hicpro环境并安装HiC-Pro(需要root权限)
conda activate HiC-Pro
make configure
make
这样是最适合小白同学的安装啦,推荐使用官方的yml文件创建环境,如果是自己创建环境下载依赖的话,记得去修改config-install.txt文件的地址哦
下面小果开始带大家一起使用HiC-Pro来分析HiC数据啦
首先我们需要的文件是参考基因组,bowtie2软件建立的索引,含有酶切片段信息的bed文件,基因组大小信息的chrom.size文件以及Hi-C的测序数据集(这里小果使用测试数据集)
首先下载测试数据集:
wget http://juicerawsmirror.s3.amazonaws.com/opt/juicer/work/HIC003/fastq/HIC003_S2_L001_R1_001.fastq.gz
wget http://juicerawsmirror.s3.amazonaws.com/opt/juicer/work/HIC003/fastq/HIC003_S2_L001_R2_001.fastq.gz
小果参考基因组选择的是hg38,接着使用bowtie2-build来建立索引:
bowtie2-build -f hg38.fasta hg38
参数-f指定参考基因组,后面是索引文件的前缀,最后会生成6个bt2文件
酶切片段信息的bed文件使用digest_genome.py来生成,digest_genome.py脚本在HiC-Pro-3.1.0/bin/utils目录下面:
python3 /HiC-Pro-3.1.0/bin/utils/digest_genome.py hg38.fasta -r MboI -o hg38_MboI.bed
参数-r指定酶切种类,不区分大小写
参数-o指定生成文件名称
小果生成的hg38_MboI.bed文件如下:
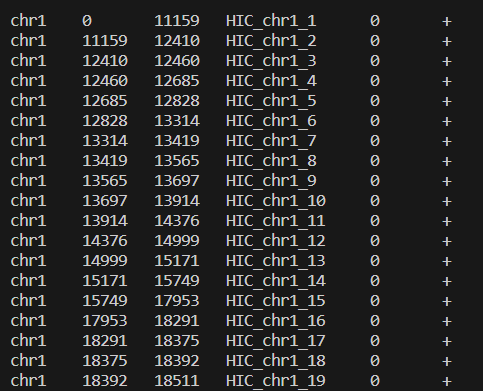
其中第三列是检测到的酶切位点对应的位置
小果使用samtools和awk工具来得到chrom.size文件:
首先使用samtools软件对参考基因组建立索引:
samtools faidx hg38.fasta
然后使用awk来对fai文件进行操作:
awk ‘{print $1 “\t” $2}’ hg38.fasta.fai > hg38.chrom.sizes
上面文件的准备工作已经完成了,下面开始使用HiC-Pro软件进行分析了
不过我们首先要在HiC-Pro的软件根目录下创建一个rawdata文件夹来存放我们的测序数据,
下面我们要修改配置文件:config-hicpro.txt,下面是一些重要的设置参数:
- N_CPU,CPU数目;
- BOWTIE2_IDX_PATH,索引所在目录
- REFERENCE_GENOME,比对参考基因组路径及前缀
- GENOME_SIZE,chrom.sizes文件的路径
- GENOME_FRAGMENT,酶切片段的bed文件的路径
- LIGATION_SITE,酶切位点末端补平再次连接后形成的嵌合序列,例如HindIII,则为AAGCTAGCTT;如果是MboI则序列为GATCGATC;
配置好文件,我们就可以跑起来啦
小果的代码如下:
YOUR_PATH/bin/HiC-Pro -c YOUR_PATH/config-hicpro.txt -i YOUR_PATH/rawdata/ -o YOUR_PATH/test_re
其中,-c参数指定配置文件,-i指定数据的路径,-o指定生成文件夹的路径
生成的日志文件在-o指定的文件夹里面
下面是小果的结果文件:
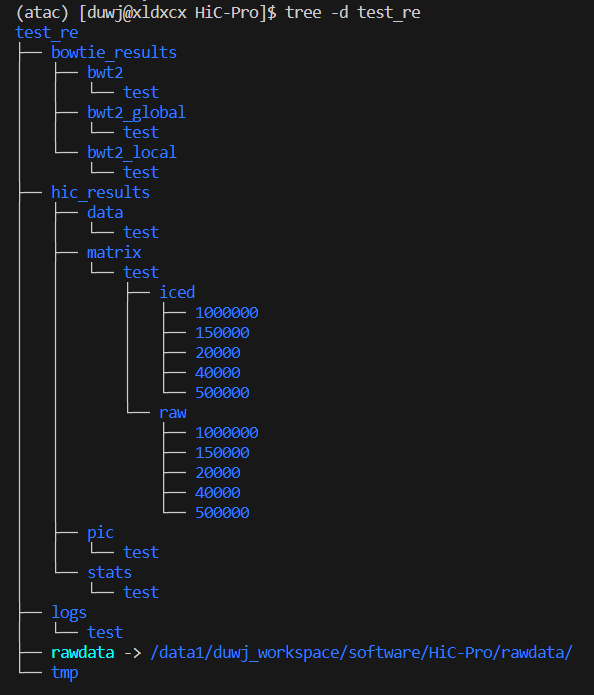
其中,bowtie_results:比对结果所在目录;
hic_results:hic矩阵及分析结果所在目录;
logs:存放分析日志;
rawdata:链接了原始数据;
tmp:存放中间文件
目录bowtie_results下共有三个文件夹:
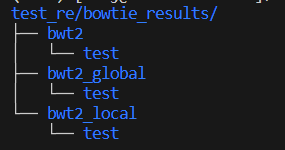
- bwt2:存放合并后的bam文件和统计结果
- bwt2_global:存放全局比对结果
- bwt2_local:存放局部比对结果
目录hic_results下面有四个文件夹:
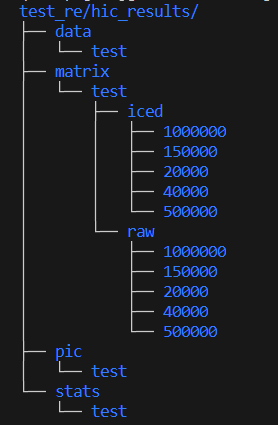
- data:存放valid pair reads及其他数据文件
- matrix:存放不同分辨率矩阵文件
- pic:存放统计分析图片
- stats:存放统计表
今天的关于HiC-Pro软件的学习就到这里啦,感兴趣的小伙伴可以找小果讨论,有感觉生信分析复杂的小伙伴可以直接使用我们的生信小工具哦,链接在这:http://www.biocloudservice.com/home.html,我们明天见咯~