大家好,小果又跟大家见面啦!这次小果给大家分享三维基因组的AB区室分析的内容
以防小伙伴们跟不上小果的节奏,小果首先跟大家介绍一下三维基因组的一些相关概念:
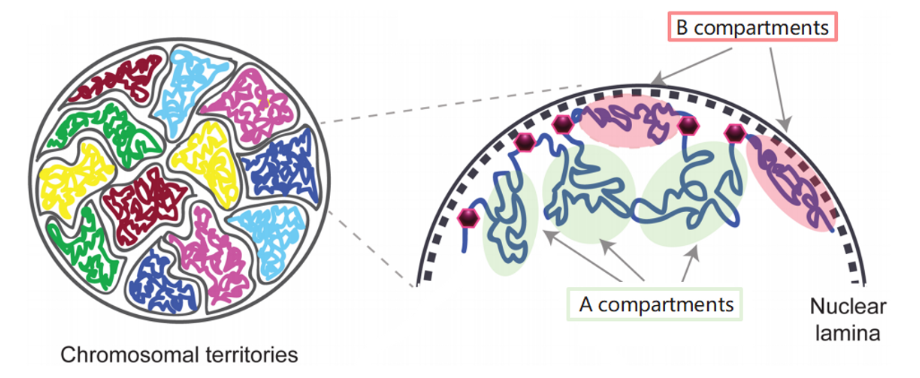
- 在真核生物的基因组中,细胞核内的染色质分布并不是随机的,为了跨越较大的基因组的距离去相互作用,例如增强子和启动子的互作,这些密切作用的染色质会靠的更近,这就是染色质疆域,如下图所示:

- 整个基因组被分割为两个空间区室,分别标记为A,B染色质区室,往往区室内互作频繁,而区室间互作较少。在生物信息分析中,通常通过计算染色体内部互作的相关性来区分两种不同的区室。其中A区室是开放的染色质,表达活跃,基因丰富,具有较高的CG含量,包含用于主动转录的组蛋白标记,通常位于细胞核内部;而B区室是关闭的染色质,表达不活跃,基因缺乏,结构紧凑,含有基因沉默的组蛋白标志物,位于细胞核的外围。AB区室示意图如下所示:
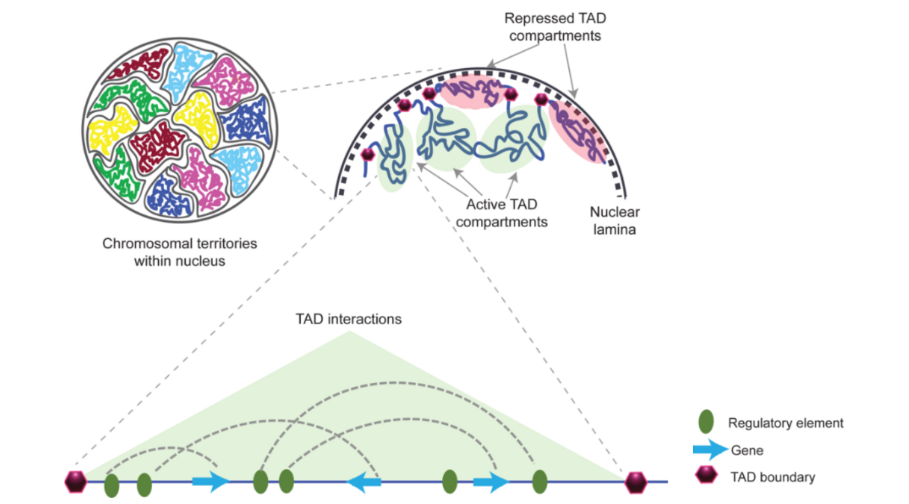
- 在染色质区室中,还存在互相作用相对频繁的基因区域,被称为拓扑结构域(TAD)。这些区域在不同的哺乳动物的不同细胞中都很保守,并且高度富集CTCF和粘附蛋白。TAD结构如下图所示:
上面带大家了解了部分三维基因组的知识,下面小果带大家进行三维基因组的AB区室分析,发车咯
目前比较流行的计算AB区室的软件主要有一下三种:
- juicer: 第一篇HiC文章实验室提供的工具,计算多个分辨率每个bin的PC1值,但是输出只有一列,不是bedgraph文件,不太人性化;同时不能判断AB区室,需要自己手动校正。
- homer: NGS分析套件,也有HiC分析流程,可以通过TSS或者ATAC测序数据进行判断AB区室,也可以输出bedgraph文件和矩阵文件。
- cworld: 一种基于Perl的HiC分析工具,可以计算AB区室和TAD等结构。
上述三个软件的优缺点如下:
- juicer: 优点是第一篇HiC文章实验室提供的工具,有权威性和可靠性,可以计算多个分辨率每个bin的PC1值;缺点是输出只有一列,不是bedgraph文件,不太人性化,同时不能判断AB区室,需要自己手动校正。
- homer: 优点是NGS分析套件,也有HiC分析流程,可以通过TSS或者ATAC测序数据进行判断AB区室,也可以输出bedgraph文件和矩阵文件;缺点是安装配置比较复杂,需要下载或者构建基因组信息。
- cworld: 优点是一种基于Perl的HiC分析工具,可以计算AB区室和TAD等结构,也可以输出矩阵文件;缺点是文档不太完善,使用不太方便。
大家了解了这三个软件的区别之后,是不是感觉homer软件更全面呢,反正小果选择的是HiC-Pro+homer来进行AB区室计算的。
首先需要用到的是HiC-Pro软件生成的.allValidPairs文件,没有使用过HiC-Pro软件的小伙伴可以去看以前小果的文章嗷。
下面是格式的转换,修改.allValidPairs文件格式,将HiC-Pro输出文件,转换成homer格式:
cat $test.allValidPairs | awk ‘BEGIN{FS=OFS=”\t”}{print NR,$2,$3,$4,$5,$6,$7}’ > test.homer
下面是homer软件的安装:
HOMER的安装可以使用conda来完成:
conda install -c bioconda homer
下面需要configureHomer.pl完成Homer软件的配置:
- 先下载configureHomer.pl:
wget http://homer.ucsd.edu/homer/configureHomer.pl
- 使用configureHomer.pl配置Homer(小伙伴们注意文件路径问题嗷)
perl configureHomer.pl [options]
大家可以使用configureHomer.pl来配置Homer,例如下载人类,小鼠的参考基因组等
万事具备,只欠东风。下面小果开始分析,小果的代码是这样的:
## 第一步 makeTagDir
# -format指定格式,test.homer为输入的homer格式文件
makeTagDirectory test -format HiCsummary test.homer
## step 2 : calculate PC1 value
# test-500k为生成文件前缀,-res是以bp为单位的分辨率,-genome为参考基因组
runHiCpca.pl test-500k test -cpu 32 -res 500000 -genome hg38.fasta -pc 1
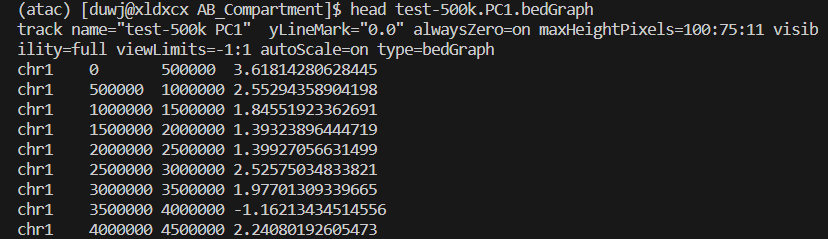
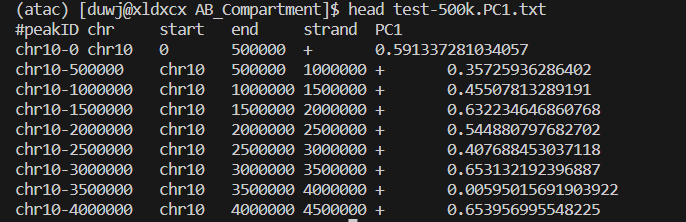
结果会生成两个文件,分别是bedGraph文件txt文件
小果的bedGraph文件是这样的:
Txt文件是这样的:
两个文件的内容大致相同,只是格式不同,bedGraph文件可使用IGV软件进行可视化,小果的可视化结果如下图:
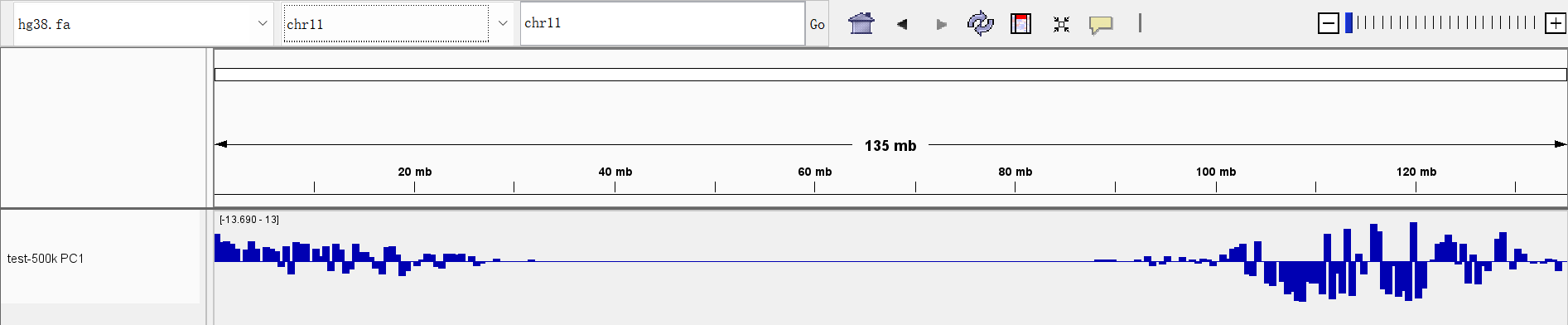
以11染色体为例,大家可以清楚的看到染色体被分为AB两个区室(以PC1值划分)
今天的关于三位基因组AB区室的计算学习就到这里啦,感兴趣的小伙伴可以找小果讨论,有感觉生信分析复杂的小伙伴可以直接使用我们的生信小工具哦,链接在这:http://www.biocloudservice.com/home.html,我们明天见咯~