Hi!小果今天来教大家一个探索基因组信息的得力助手工具,那就是我们今天的主角:R语言中的GenomicRanges包,接下来小果将为大家介绍GenomicRanges包基本功能和用途,以及如何实战应用,感兴趣的话就和小果一起看下去吧!
什么是GenomicRanges?
“GenomicRanges”是一个在R语言中广泛使用的包,它提供了丰富的功能和工具,用于处理和分析基因组范围数据。该包主要用于处理基因组中的基因、转录本、染色体等生物学特征的位置和范围信息。它可以帮助研究人员在基因组水平上进行注释、可视化和分析,从而深入理解基因组的结构和功能。 “GenomicRanges”包在生物信息学和基因组学领域具有重要的应用,常用于以下任务:
- 基因注释:通过提供基因、转录本和外显子的位置信息,”GenomicRanges”可以帮助研究人员对基因进行注释,了解基因的结构和功能。
- 基因组比对和对比:该包可以帮助研究人员在不同基因组之间进行比对和对比,识别共有的或差异的基因区域,以及进行基因组重排等分析。
- 基因组范围的操作和计算:“GenomicRanges”提供了丰富的函数和方法,用于基因组范围的操作,如合并、分割、平移、过滤、排序等。此外,还可以进行基因区域的计算,如覆盖率、交集、差集等。
- 可视化:“GenomicRanges”包可以与其他数据可视化包(如ggplot2、Gviz等)结合使用,帮助研究人员绘制基因组范围的图形和图表,展示基因的位置、分布和特征。
安装及加载R包
在这里小果给大家提供两种安装方法,大家根据自己R语言的版本来自行选择哦!
install.packages(“GenomicRanges”) #安装方法一
BiocManager::install(“GenomicRanges”) #安装方法二
library(GenomicRanges)
GenomicRanges对基因组数据进行分析
现在,”GenomicRanges”包已经成功安装和加载,可以开始使用它进行基因组范围数据的处理和分析。
首先,我们需要加载”TxDb.Hsapiens.UCSC.hg38.knownGene”包。这是一个包含人类基因组hg38版本的转录本数据库,可以用于基因注释和相关信息。 我们将针对该数据集进行一系列的分析:
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
接下来,我们将使用TxDb.Hsapiens.UCSC.hg38.knownGene对象创建一个名为txdb的数据库对象,并从中获取hg38Gene基因信息。
hg38Gene <- genes(txdb)
- 了解对象类型
我们可以使用class()函数来查看对象的类型:
class(hg38Gene)
- 显示对象结构
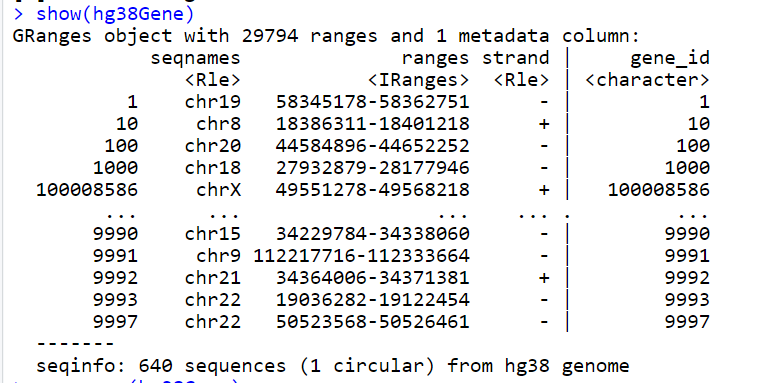
我们可以使用show()函数来显示对象的结构:
show(hg38Gene)
- 查看指定信息列
- 查看seqnames信息列,使用seqnames()函数来查看hg38Gene对象中的seqnames信息列:
seqnames(hg38Gene)

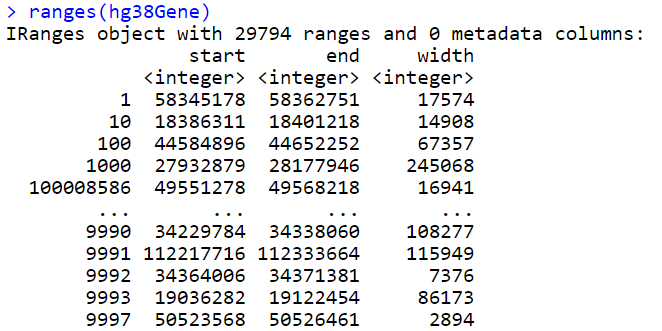
2.查看ranges信息列,使用ranges()函数来查看hg38Gene对象中的ranges信息列:
ranges(hg38Gene)
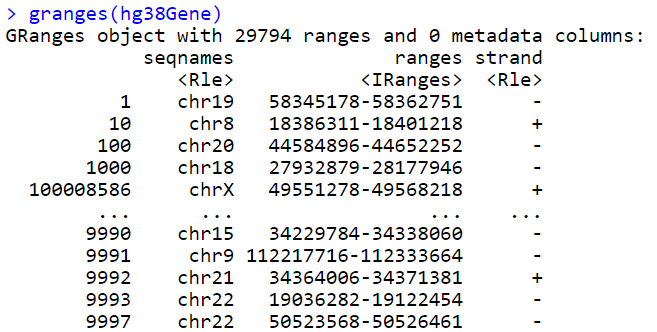
3.提取左边信息。使用granges()函数来提取hg38Gene对象中的左边信息。
granges(hg38Gene)
- 分割与和合并对象
- 分割GRanges为多个对象。我们可以使用split()函数将hg38Gene对象分割为多个对象。在这个例子中,我们将对象分割为两个部分:
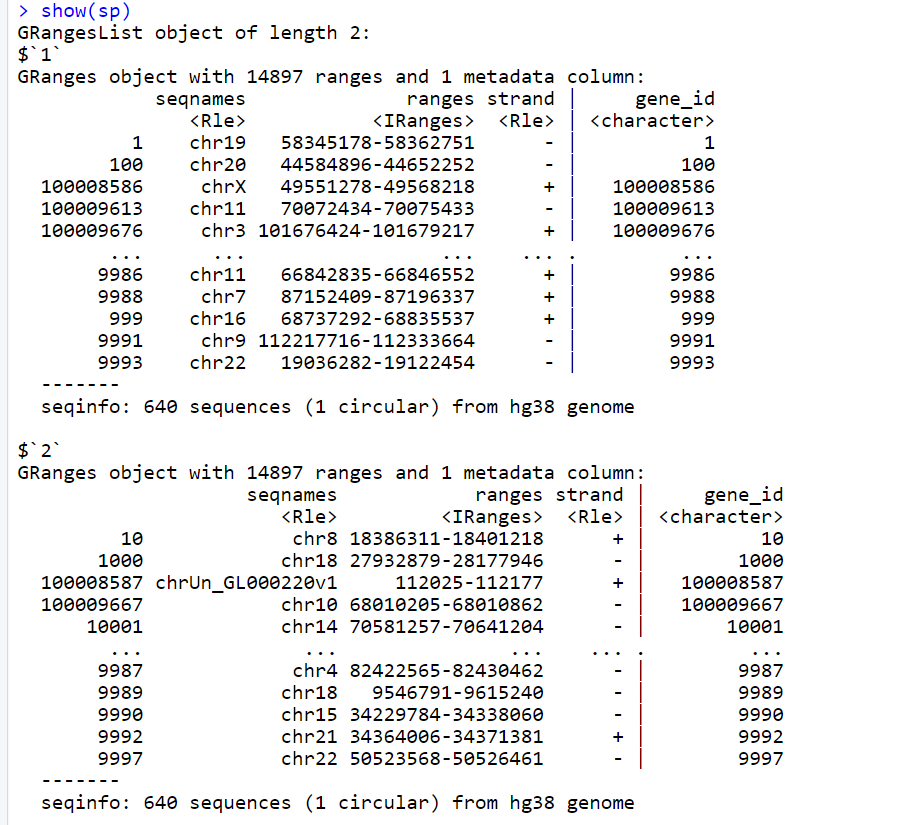
sp <- split(hg38Gene, rep_len(1:2, length.out = 25750))
show(sp)
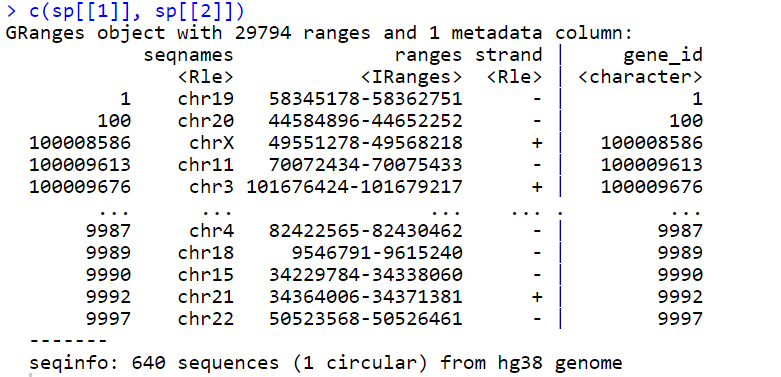
- 合并多个对象。使用c()函数来合并多个对象。在这个例子中,我们将sp[[1]]和sp[[2]]对象合并。
c(sp[[1]], sp[[2]])
- 移动基因区间
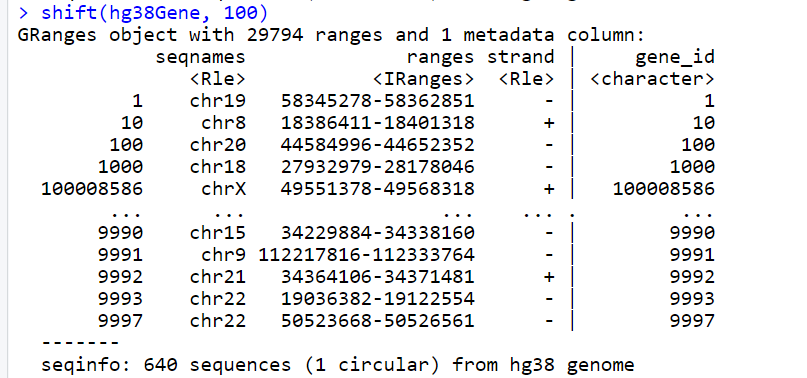
- 对每一个基因区域移动100bp。使用shift()函数来将每个基因区域向右侧移动100个碱基对:
shift(hg38Gene, 100)
- 统计区域重叠情况。使用coverage()函数来统计hg38Gene对象中基因区域的重叠情况。
coverage(hg38Gene)

以上代码就是对GenomicRanges包的介绍与基本使用方法,学会这些方法后,你可以学习如何使用GenomicRanges包进行基因组分析,包括创建数据库对象、获取基因信息,以及对基因区域进行操作和统计等。当然,大家也可以根据自己的需求和研究目标修改和扩展这些代码哦!!怎么样,你学会了嘛!?