首先,什么是基因家族呢?所谓的基因家族就是一类拥有类似结构,行使相同功能的蛋白家族,把这个结论放在核算序列或者氨基酸序列层面上,就是可能具有极为保守的功能域,而导致这类蛋白具有类似的结构和行使相同的功能。
基因家族分析通常由以下4个步骤组成:
1 基因家族成员的鉴定
(这是最关键、最重要的一步,也是小果认为最有用的一个环节,需要谨慎分析,这一步出现问题,后面的分析也会有较大偏差。);
1.1 基因组文件(dna, cds, protein, gff3 or gtf注释文件)的获取;
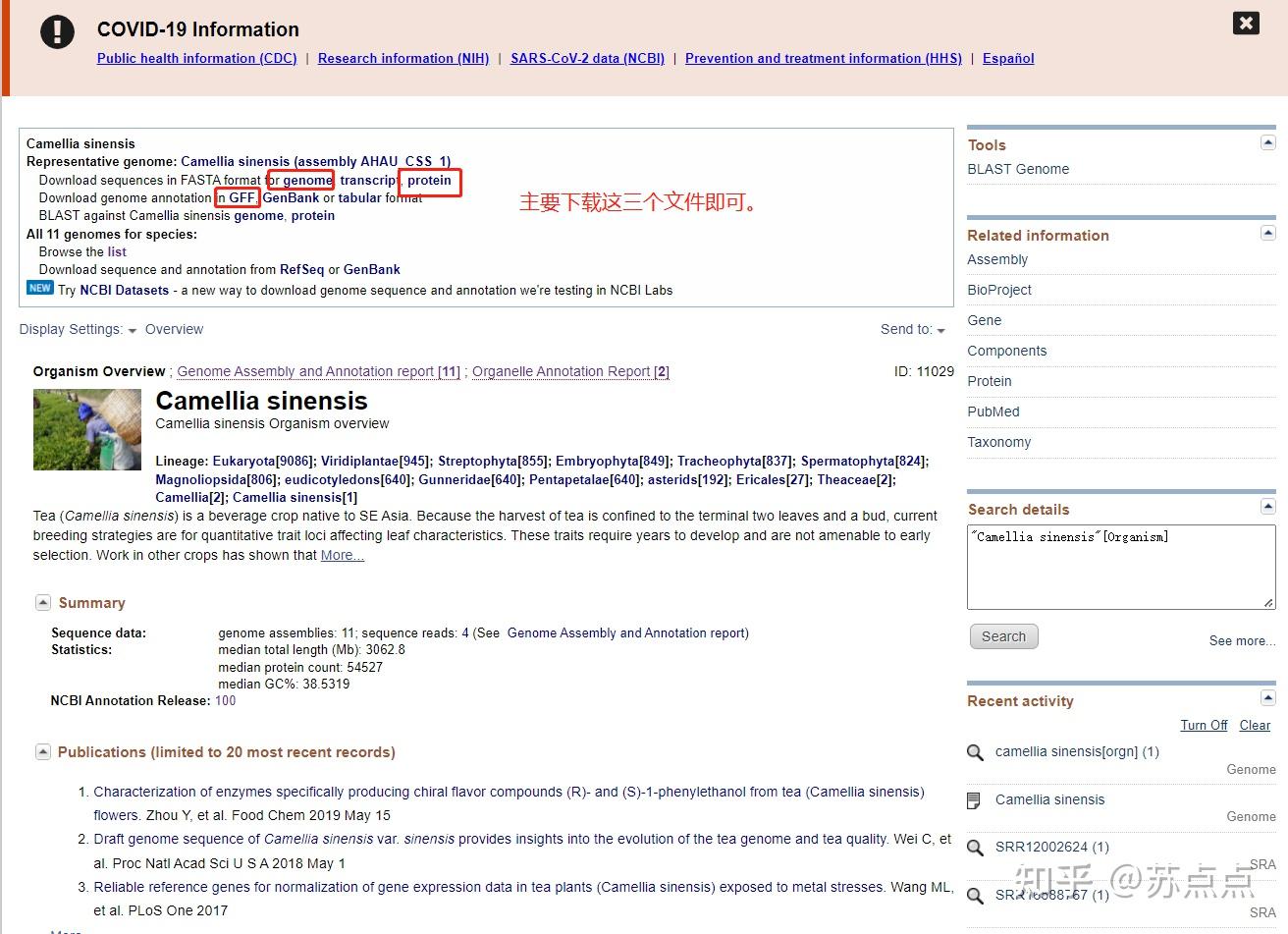
1.2 基因家族隐马尔可夫模型(HMM model)的获取;
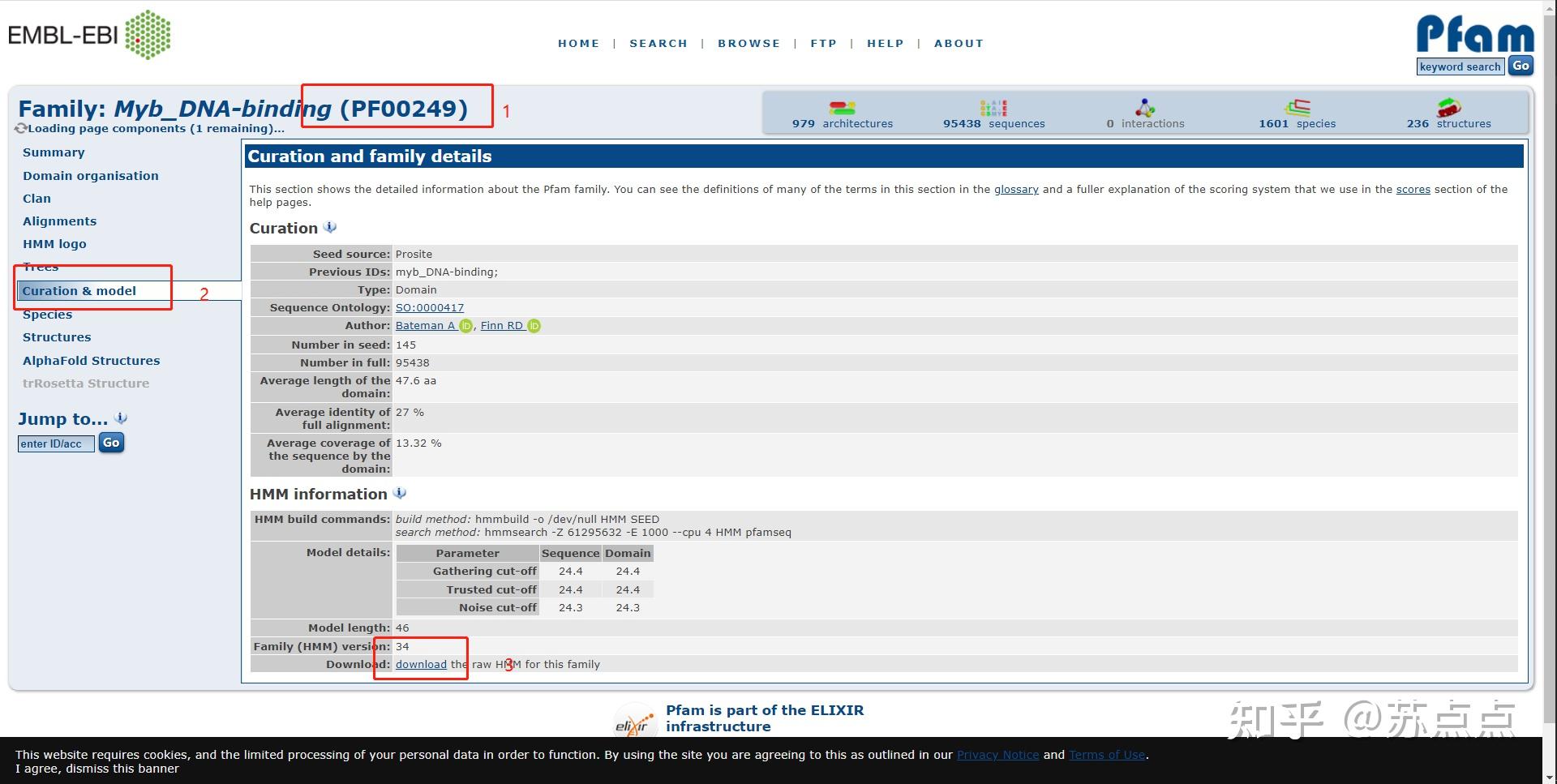
1.3 若没有对应的HMM model,则需要自己构建;
1.4 通过HMM model在基因组文件中搜寻 候选基因家族成员;
1.5 通过本地blast+获取参考基因家族的同源序列 (候选基因家族成员);
1.6 获取候选基因家族成员的序列信息;
1.7 结合系统发育树、保守结构域和保守基序(motif)等确定最终的基因家族成员;
1.8 检查注释文件是否正确。
2 基因家族成员序列基本信息的获取
(简单操作,实际上很多结果可能用不到,会在实验设计过程中起到一定的参考。)
2.1 基因家族成员蛋白的理化性质;
2.2 跨膜结构域、信号肽、亚细胞定位等;
3 构建系统发育树
(系统发育树是一个较大的版块,需要掌握的内容还是蛮多的。单就构建系统发育树来说,相对简单,一般也容易操作。但在树的可视化及美化编辑过程中往往并不那么容易,因为树可以呈现的内容可以非常丰富,至于如何展示,真的全在个人。这里先不展开。)
好吧,废话到此为止。
下面系统的整理一下小果在基因家族分析流程中的全部的实操流程及一些思考。
一、基因家族成员的鉴定
#step1 在Pfam数据库中下载基因家族蛋白保守结构域的隐马尔可夫模型(.hmm)
#step2 在ensembl数据库中下载物种的基因组序列文件,CDS序列文件,gtf基因组注释文件,pep蛋白质序列文件
Homo_sapiens.GRCh38.dna.toplevel.fa.gz
Homo_sapiens.GRCh38.cds.all.fa.gz
Homo_sapiens.GRCh38.pep.all.fa.gz
Homo_sapiens.GRCh38.98.gtf.gz
gata.hmm
#step3 利用HMMER软件初步鉴定基因家族成员(一般利用 –cut——tc算法)
cd /home/GeneFamily/DATA/human (此路径就是上述数据下载位置)
/home/GeneFamily/TOOLS/hmmer/binaries/hmmsearch –cut_tc –domtblout gata.out /gata.hmm Homo_sapiens.GRCh38.pep.all.fa
此步会生成初步的结果gata.out文件
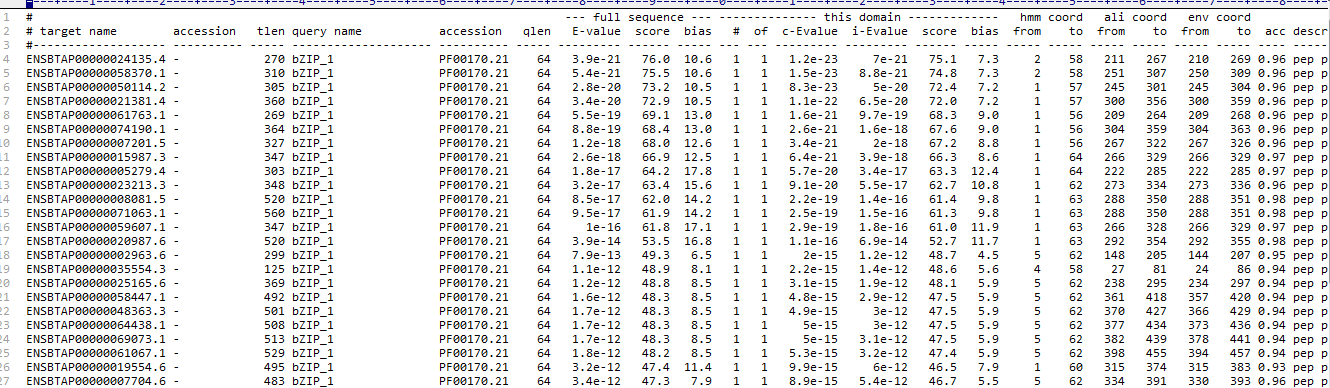
#step4 对gata.out文件初步质控,筛选e值小于1*10^-20的结果
grep -v “#” gata.out|awk ‘($7 + 0) < 1E-20’|cut -f1 -d ” “|sort -u > gata_id.txt
#step5 利用Samtools软件把筛选后的蛋白质序列挑出来
首先利用samtools软件对pep文件生成索引文件
/home/NGS/tools/samtools/samtools faidx Homo_sapiens.GRCh38.pep.all.fa
会生成一个名为Homo_sapiens.GRCh38.pep.all.fa.fai 的文件
然后利用samtools软件挑出序列
xargs /home/NGS/tools/samtools/samtools faidx Homo_sapiens.GRCh38.pep.all.fa < gata_id.txt > gata_protein.fasta
但是生成的fasta文件是含有回车符的所以利用一个见得perl命令消除回车符
perl -pe ‘/^>/ ? print “\n” : chomp’ gata_protein.fasta | tail -n+2 > gata_protein_1.fasta
#step6 利用clustalX软件对生成的fasta文件多重序列比对,结果会生成gata_protein_1.aln文件
#step7 利用hmmbuild命令对置信序列进行隐马尔可夫模型的构建,精准预测所有基因家族成员
/home/GeneFamily/TOOLS/hmmer/binaries/hmmbuild gata_build.hmm gata_protein_1.aln
会生成一个gata_build.hmm文件备用
然后利用hmmsearch命令精准预测
/home/GeneFamily/TOOLS/hmmer/binaries/hmmsearch –cut_tc –domtblout gata_second.out gata_build.hmm Homo_sapiens.GRCh38.pep.all.fa
会生成一个gata_second.out文件
#step8 最后再次利用e值进行过滤
grep -v “#” gata_second.out|awk ‘($7 + 0) < 1E-3’|cut -f1 -d ” “|sort -u > gata_fina_id.txt
xargs /home/NGS/tools/samtools/samtools faidx Homo_sapiens.GRCh38.pep.all.fa < gata_fina_id.txt > gata_fina_protein.fasta
二、利用PhyML对上述得到的成员的序列进行极大似然树的构建
#step1 利用prottese软件选择建树的最佳进化模型
java -jar prottest.jar -i gata_protein_1.phy -o gata_good_model.txt -S 1 -AIC -I -G -IG -F -ncat 8 -all
其中-i:输入文件(.phy格式),-o:输入的最佳模型的结果文件
-S:运算的策略,fast为0,slow为1,但是slow会更精准一些
-AIC:用于排序的framework 除了AIC之外还有AICc BIC DT 等等
-G -I -IG -F:为几种构建模型的策略,在做model的时候,全选上
-ncat 8指的是8个rate category, phyML建树时也会设置这个参数,在一定范围内越大越好,但是不能太大
-all 汇总各个framwork的排序结果
这一步会生成一个.txt的结果文件,deltaAIC值为0或者最小的时候,该模型是最佳模型,如下:
***************************************************************************
Best model according to AIC: JTT+I+G+F
Confidence Interval: 100.0
***************************************************************************
Model deltaAIC AIC AICw -lnL
—————————————————————————
JTT+I+G+F 0.00 12690.92 1.00 6285.46
VT+I+G+F 18.27 12709.19 0.00 6294.59
JTT+G+F 30.07 12720.99 0.00 6301.50
然后在文件的上方找到这个模型和它的所有参数。如下
Model………………………….. : JTT+I+G+F
Number of parameters…………… : 60 (21 + 39 branch length estimates)
gamma shape (8 rate categories).. = 2.298
proportion of invariable sites… = 0.139
aminoacid frequencies………… = observed (see above)
-lnL………………………….. = 6285.46
(seconds))
其中gamma shape (8 rate categories).. = 2.298
proportion of invariable sites… = 0.139这两个参数在之后的phyml软件建树的构成中需要用到。
到这最佳模型的选择就完成了,加下来利用PhyML进行极大似然树的构建
#step2 利用phyml构建极大似然树
此步见(PhyML利用氨基酸序列建树.pdf)文件
以上是基因家族分析的基本流程,当然基因家族分析的内容不只这些,小伙伴们可以再进一步分析这个预测蛋白的结构域(CD-Search)、这个基因在染色体上的位置、重复事件、共线性分析、SNP等内容。进阶版教程可以期待一下。
有疑问欢迎咨询小果的微信公众号(生信果)和云生信生物信息学平台( http://www.biocloudservice.com/home.html)