在这一篇中,小果将带大家学习如何去使用Mega软件做多序列的比对和构建基因进化树。
首先,先和大家介绍下。MEGA(Molecular Evolutionary Genetics Analysis)软件是一种常用的生物信息学工具,主要用于分析和推断生物序列(如DNA、蛋白质序列)的进化关系和亲缘关系。通过构建进化树,可以帮助研究人员了解不同生物种群之间的演化历程和分类关系。那么接下来就开始比对和建树吧!
一、多序列比对
首先,我们现在mega的首页选择”ALIGN”,点击 “Edit/Build Alignment” ,会弹出一个对话框,选择”Create a new alignment”,根据需要比对的序列 (氨基酸序列或核苷酸序列),选择”DNA”或”Protein”。由于我们上一篇下载的数据为Synaptophysin蛋白序列,所以我们可以选择Protein。
紧接着,我们可以点击”Edit”,选择”Insert Sequence From File”导入我们需要比对的序列,序列文件格式为.fasta格式。
打开在上一篇中下载的sequence.fasta,导入到mega软件中,可以得到的序列情况如下所示: 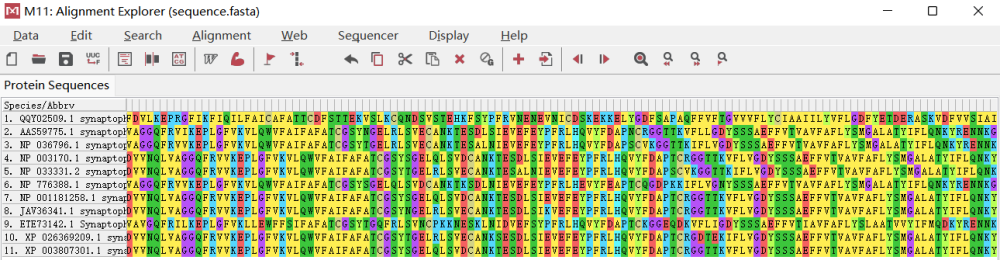
接着我们要对目标序列进行多序列比较,我们选择ClustalW,调整参数(一般用默认参数),即可完成多序列比对。

然后我们可以再看到序列这边,为了使进化树不会构建失败,我们需要删掉两端差异较大的地方,留下基因保守的地方,使得两端对齐。因为如果序列的相似性太差的话,进化树就很可能构建失败。具体的步骤就是看看序列两端的对齐情况是否差异性较大,如图所示我们将这段删除。先选中此段序列,点击鼠标右键,选择”Delete”。

紧接着,我们点击”Data”,选择”Phylogenetic Analysis”进行系统发育分析。

二、基因进化树的构建
接着,我们返回主页面,点击”PHYLOGENY”,构建系统发育树主要有三种方法,分别是最大似然法 (Maximum Likelihood)、邻接法 (Neighbor-Joining) 和最小进化法 (Minimum Evolution)。 其中:
- 最大似然法(Maximum Likelihood): 最大似然法是基于概率统计原理的一种方法。它通过计算给定数据集的多个进化树模型中最大化似然函数的树结构,从而找到最可能的进化树。最大似然法考虑了每个进化树模型中观察到的序列数据出现的概率,并根据最佳拟合的模型参数进行优化。
- 邻接法(Neighbor-Joining): 邻接法是一种启发式的方法,它基于序列数据的相似性来构建进化树。邻接法首先计算序列之间的距离矩阵,然后根据距离矩阵中的最小距离,将序列一对一对地合并为一个节点,直到构建出一棵完整的进化树。邻接法适用于较大的序列数据集,计算速度相对较快。
- 最小进化法(Minimum Evolution): 最小进化法是一种基于树长度的方法。它尝试寻找一棵进化树,使所有序列之间的进化步骤总和最小。最小进化法通过优化树的拓扑结构和分支长度来求解最小进化树。这种方法在序列数据短小且进化关系较简单的情况下效果较好。
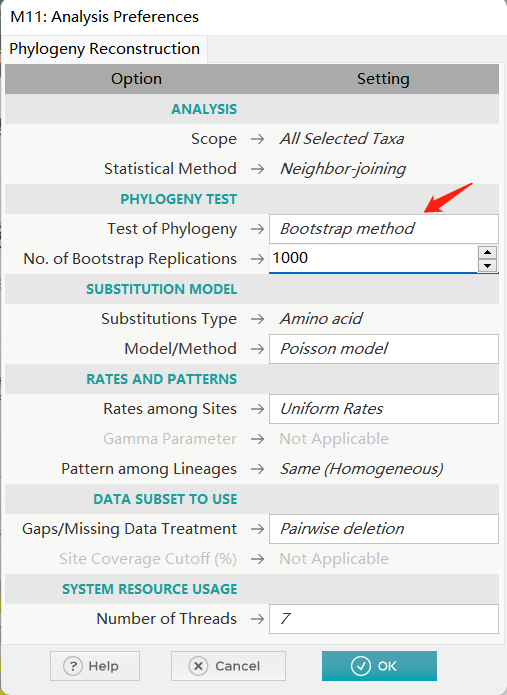
为了能够更快地得到匹配结果,我们采用邻接法,其中的方法将选用Bootstrap method。因为邻接法通常与Bootstrap方法结合使用,以评估构建的进化树的可靠性。Bootstrap方法是一种重采样技术,用于生成多个数据集的重复样本,从而通过多次构建进化树来估计结果的可靠性。
最终得到的基因进化树结果如下:
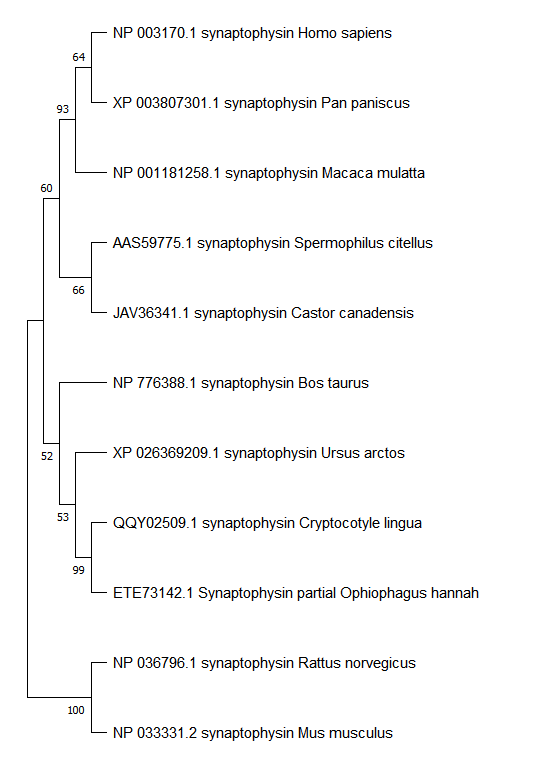
相信通过这篇的讲解,大家也会对基因进化树的构建软件的使用有了大概的了解了吧!接下来可以自己实操一下,然后对基因进化树进行构建!如果大家还对其他方面的生信工具感兴趣的话,欢迎大家点击http://www.biocloudservice.com/home.html来进行学习哦!