Trinity, 是由Broad Institute开发的进行de novo转录本组装的工具,它能将RNA-seq reads拼接成更长的转录本。Trinity由三个模块组成,其基本作用如下所示:
- Inchworm:将RNA-seq reads组装成单个转录本
- Chrysails:将上一步形成的转录本进行聚类,对于每一个聚类形成完整的de Bruijin graphs。每一个聚类代表一个基于转录本的复杂程度。
- Butterfly:并行的处理每一个图,追踪图中reads和配对reads的路径,最后报道可变剪切亚型的全长转录本。
快来跟着小果一起学习吧!
- 软件安装
Trinity需要在Linux系统上运行。
软件下载链接:https://github.com/trinityrnaseq/trinityrnaseq/releases 
我们打开可以发现该软件开发的很早,但仍然能保持每年更新的状态,是非常难得的!我们选择最新版本进行下载,也就是图中的trinityrnaseq-v2.15.1.FULL.tar.gz。
下载完成后我们使用如下命令进行解压安装:
$ tar zxvf trinityrnaseq-v2.15.1.FULL.tar.gz
$ cd trinityrnaseq-v2.15.1
$ make
或者使用conda直接下载安装:
$ conda install -c bioconda trinity
- Trinity使用方法
Trinity的主要参数如下所示:
# –seqType <string>: fast文件类型(fa或者fq)
# –max_memory <string>: 使用Trinity软件时允许的最大内存
# 当数据为paired reads时使用: –left <string>: 左端reads –right <string>: 右端reads
# 当数据为unpaired reads时使用: –single <string>: 文件名
# –SS_lib_type <string>: 特异性文件文库需要添加此参数,对于paired reads有两种参数(RF、FR);对于unpaired reads也有两种(F、R)
# –CPU <int>: 指定CPU的使用个数
跟着小果一起来看一个例子吧!
运行一下代码:
$ Trinity –-seqType fq –-max_memory 10G –-left reads.left.fq.gz –-right reads.right.fq.gz –SS_lib_type RF –CPU 4
运行结束时会自动生成一个trinity_out_dir的文件夹,里面记载着所有输出文件,如‘trinity_out_dir.Trinity.fasta’, 其中一个fasta条目如下所示:
>TRINITY_DN1000_c115_g5_i1 len=247 path=[31015:0-148 23018:149-246]
AATCTTTTTTGGTATTGGCAGTACTGTGCTCTGGGTAGTGATTAGGGCAAAAGAAGACAC
ACAATAAAGAACCAGGTGTTAGACGTCAGCAAGTCAAGGCCTTGGTTCTCAGCAGACAGA
AGACAGCCCTTCTCAATCCTCATCCCTTCCCTGAACAGACATGTCTTCTGCAAGCTTCTC
CAAGTCAGTTGTTCACAGGAACATCATCAGAATAAATTTGAAATTATGATTAGTATCTGA
TAAAGCA
‘TRINITY_DN1000_c115‘是聚类的标号,’g5‘代表一个基因,’i1‘代表一个转录本。
因为是算法帮助我们组装的转录本,我们怎么知道它组装的好坏呢?接下来小果将继续介绍如何用不同方法查看转录本的组装质量。
- 计算全长转录本数量
其中一种用来衡量拼接质量的方法就是数全长或者接近全长转录本的数量,越多代表组装的效果越好。对于模式物种,我们可以将转录本比对到参考集上去,而对于非模式物种,如果存在有高质量注释的近源物种,我们可以使用它们的参考集进行衡量。
我们使用Blast+进行分析,首先构建蛋白质数据库
$ makeblastdb -in uniprot_sprot.fasta -dbtype prot
然后进行搜索
$ blastx -query Trinity.fasta -db uniprot_sprot.fasta -out blastx.outfmt6 -evalue 1e-10 -num_threads 6 -max_target_seqs 1 -outfmt 6
得到了输出文件之后,我们可以使用Trinity软件之中的脚本自动帮我们检测Trinity转录本比对到参考组上的百分比
$ TRINITY_HOME/util/analyze_blastPlus_topHit_coverage.pl blastx.outfmt6 Trinity.fasta uniprot_sprot.fasta
跑完之后我们会得到一个’blastx.outfmt6.txt.w_pct_hit_length’文件,通过它我们可以画出一个表格来更好的展示结果,如下:
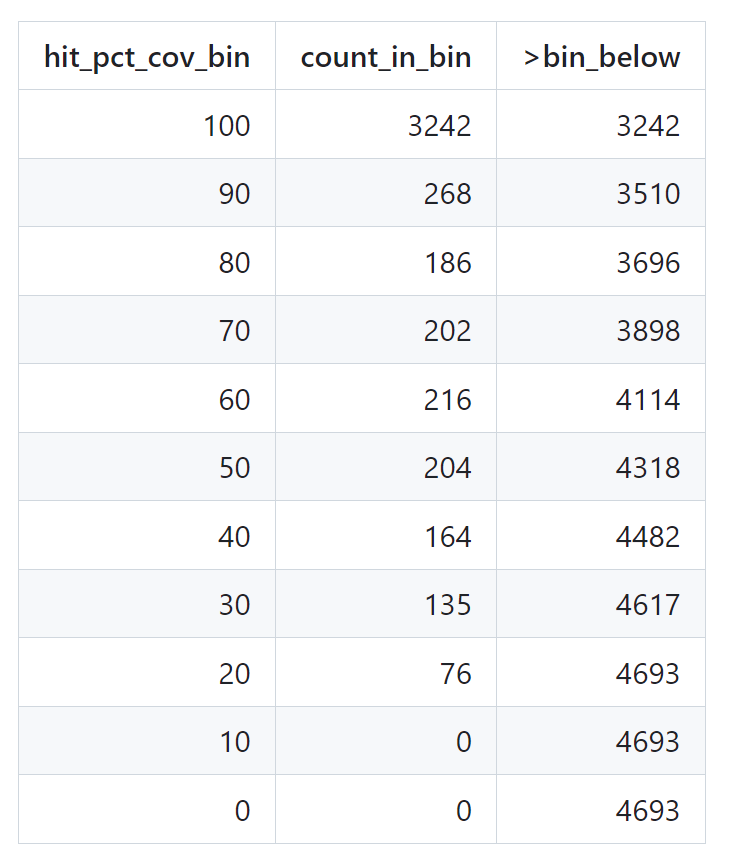
看到图表中的第二行,其含义为数据库中有268个蛋白质与Trinity转录本匹配,匹配长度为每个转录本长度的80-90%;数据库中有3510个蛋白质可以被几乎为全长的转录本表示,比对覆盖度大于80%。第一行的信息为,有3242个蛋白质与转录本匹配,匹配长度均大于90%。
- RNA Seq Read Representation
另外一种方法是为了检测RNA seq reads能否较好的表示Trinity组装的转录本,为了全面捕获reads,我们使用Bowtie2将reads和转录组对齐,然后计算正确的不正确的和被孤立的reads的数量
首先,我们为转录本建立索引
$ bowtie2-build Trinity.fasta Trinity.fasta
然后进行比对来获取read的比对信息, 我们以双端数据为例
$ bowtie2 -p 10 -q –no-unal -k 20 -x Trinity.fasta -1 reads_1.fq -2 reads_2.fq 2>align_stats.txt| samtools view -@10 -Sb -o bowtie2.bam
最后可以查看输出文件’align_stats.txt’
$ cat 2>&1 align_stats.txt
结果如下所示: 
我们可以看到有89.97%的比对率,总的来说对于双端数据,有70~80%的比对率就算合格了。
好了今天的分享就到这里啦~有什么问题可以和小果一起讨论哦~下一期小果继续为大家带来Trinity的更多后续内容哦
更多生信小工具可查询云平台:http://www.biocloudservice.com/home.html