生存分析是一种统计方法,用于分析个体在给定时间段内发生某个事件(如死亡)的概率或时间。生存分析可用于评估不同因素对个体生存时间的影响,确定生存率曲线,比较不同组之间的生存差异,并预测患者的生存期。常见的生存分析方法包括Kaplan-Meier方法、Cox比例风险回归分析和生存ROC曲线分析。
Kaplan-Meier方法(Kaplan-Meier analysis):Kaplan-Meier方法用于估计生存函数,即给定时间点上生存的概率。它可以根据不同的组别(例如不同的治疗组)绘制生存曲线,并通过对曲线进行比较来评估不同因素对生存的影响。
Cox比例风险回归分析(Cox proportional hazards regression analysis):Cox比例风险回归分析用于评估多个因素对生存时间的影响,同时控制其他因素。它可以估计各个因素的风险比(hazard ratio),即不同组之间生存风险的比较。
生存ROC曲线分析(Survival ROC curve analysis):生存ROC曲线分析用于评估生存预测模型的准确性。它通过计算生存时间的预测准确度来衡量模型的性能,可以帮助选择合适的生存预测模型。生存分析相关的包包括survival、survminer和survivalROC等。这些包提供了丰富的函数和工具,用于进行生存分析的数据处理、可视化和模型建立。
需要注意的是,生存分析需要有包含个体生存时间和事件状态(如死亡与否)的数据。常见的数据格式是时间至事件或失访的表格,例如列出每个个体的生存时间和事件状态(0表示未发生事件,1表示发生事件)。
library(limma)
library(data.table)
library(tidyverse)
library(ggsignif)
library(RColorBrewer)
library(future.apply)
library(survival)
library(survminer)
#Get all survival files
批量处理基因表达数据和生存分析数据
files=dir(“./TG_Exp/”)
使用dir函数列出指定目录./TG_Exp/下的所有文件,并将文件名存储在files变量中。
#Batch read geneexp files 批量读取基因表达文件
###
rt=read.table(“./cli_surv.txt”,header=T,sep=”\t”,check.names=F,row.names=1)
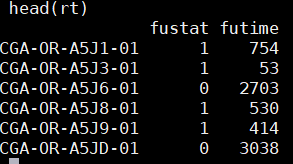
从文件cli_surv.txt中读取数据,设置文件的分隔符为制表符(\t),并将第5列和第6列的数据提取出来。然后,将这两列的列名分别设置为fustat和futime。
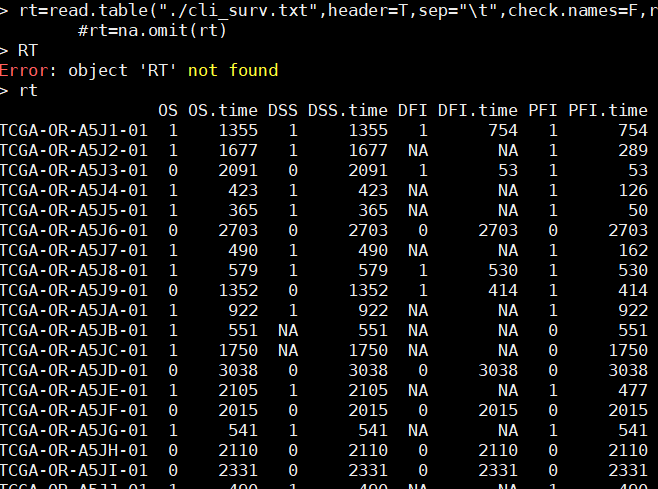
rt=na.omit(rt)
rt=rt[,c(5:6)]
colnames(rt)=c(“fustat”,”futime”)
#Batch read geneexp files
自定义的函数expTim,用于处理每个文件的基因表达数据和生存分析数据。函数的参数是一个文件路径的字符串。
在函数内部,首先读取基因表达数据文件,文件路径是由参数dirname和固定的路径字符串拼接而成。然后,使用subset函数筛选出sample_type为”tumor”的行,将第一列作为行名,并移除第一列。
接下来,通过处理dirname字符串,提取文件名部分作为变量dirnam。然后,使用intersect函数找到基因表达数据和生存分析数据中共同的样本,并将它们筛选出来。将生存分析数据和基因表达数据合并到一个数据框中,再添加一个名为”id”的列作为行名,并删除含有缺失值的行。
最后,使用write.csv函数将结果保存到指定路径下的文件中。
expTim=function(dirname){
#exp <- read.table(paste0(“./TG_GTEX_EXP/”,dirname),header=T,sep=’\t’,check.names=F)
exp <- read.table(paste0(“./TG_Exp/”,dirname),header=T,sep=’\t’,check.names=F)
exp1=subset(exp,exp$sample_type==”tumor”)
rownames(exp1)=exp1[,1]
exp1=exp1[,-1]
####
tmp<- strsplit(dirname,split=”.”,fixed=TRUE)
dirnam<-unlist(lapply(tmp,head,1))
#merging results
sameSample=intersect(row.names(rt),row.names(exp1))
surTime=rt[sameSample,]
exp1=exp1[sameSample,]
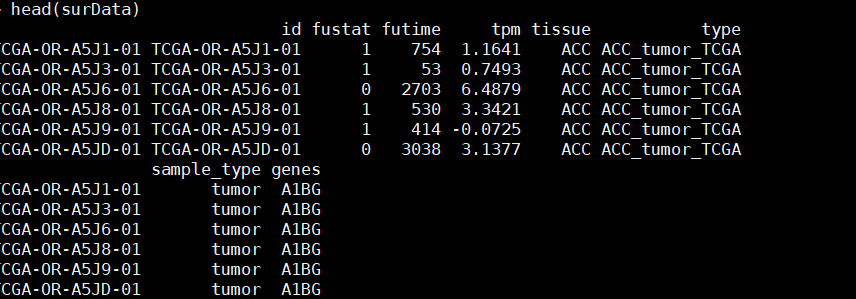
surData=cbind(surTime,exp1)
surData=cbind(id=row.names(surData),surData)
surData=na.omit(surData)
 write.csv(surData,paste0(“./DFI/”,dirnam,”_DFI_Time”,”.txt”),sep=”\t”,quote=F,row.names=F)
write.csv(surData,paste0(“./DFI/”,dirnam,”_DFI_Time”,”.txt”),sep=”\t”,quote=F,row.names=F)
}
dd=future_lapply(files,expTim)
#####
#使用future_lapply函数对files中的每个文件名调用expTim函数,并将结果存储在变量dd中。future_lapply函数是一个并行计算的版本,可以加速处理过程。后续有人想了解,可以联系小果。
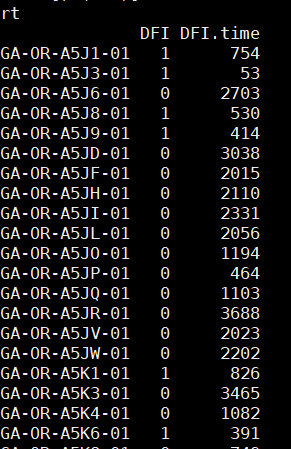
DFI(Disease-Free Interval)是指在治疗后或手术切除后,患者没有再次发生疾病的时间间隔。DFI常用于癌症领域,特别是对于评估患者的治疗效果和生存预后非常重要。DFI的计算通常涉及两个重要的时间点:
治疗开始时间(Treatment Start Time):这是指开始进行特定治疗(如手术、放疗或化疗)的时间点。疾病再发时间(Disease Recurrence Time):这是指患者再次出现疾病(如肿瘤复发)的时间点。
DFI的计算方式是从治疗开始时间到疾病再发时间的时间差。如果疾病再发事件尚未发生或数据不可用,可以将DFI视为右侧截尾(right-censored)的生存数据。
在生存分析中,DFI常用于绘制Kaplan-Meier曲线、评估治疗效果和进行生存分析。通过对患者进行随访观察,可以跟踪并记录他们的DFI,并分析不同因素对DFI的影响。
需要注意的是,DFI的计算和解释可能因研究设计、疾病类型和研究目的而有所不同。在具体的研究中,可以根据需求和特定情况对DFI进行相应的定义和分析。
下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:http://www.biocloudservice.com/home.html。
云生信平台链接:http://www.biocloudservice.com/home.html。