实验数据获得并不容易,而且成本高昂,有时只是想大致看一下几种条件的差异表达,在正式的实验前做一个预实验很有必要。同时出于成本考量,每种条件不会做重复样本。但是无重复的样本,无法使用DESeq2。但是没关系,今天小果介绍一个差异表达利器——edgeR。
包下载与安装
我们需要下载并安装两个R包,一个是edgeR,另一个是airway。airway是一个提供用于分析的数据集。
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(“edgeR”)
BiocManager::install(“airway”)
library(edgeR) #加载edgeR
library(airway) ##加载airway
构建GDElist对象
DGEList是edgeR分析差异基因中最核心的对象. 它提供两类信息: 表达量矩阵和分组信息.
为了方便大家重复,小果这里的数据来自于airway. 对于您的数据, 可以用read.table等函数进行导入.
data(“airway”)
> expr <- assay(airway)
> meta <- colData(airway)
> nrow(expr)
[1] 64102
> ncol(expr)
[1] 8
我们可以看到exp数据有64102 个基因和8个样本的矩阵
小果为了演示无重复样本,这里就用数据的前两列构建DGEList对象。
> counts <- expr[,1:2]
> group <- 1:2
> dge <- DGEList(counts=counts, group = group)
数据过滤
再一次转录过程中不可能所有的基因都有表达,因此需要预先过滤掉没有表达的基因。
> keep <- rowSums(cpm(dge)>1) >= 1
> dge <- dge[keep, , keep.lib.sizes=FALSE]
> nrow(dge)
[1] 14756
至少有一个表达的基因(CPM > 1)。,去掉全部是0,没有表达的基因。基因数就从64102降到14756了。
标准化
避免文库大小不同导致的分析误差。我们需要对数据进行标准化。
y <- calcNormFactors(y)
calcNormFactors这个函数使用的TMM(trimmed mean of M-values)来进行标准化。
无重复差异表达分析
处理好数据之后终于来到本文的重头戏了。
使用各种差异表达分析工具的目的都是为了计算)样本组间的dispersion(离散值),有了离散值之后才能能算出p值。
在无重复样本的条件下,edgeR计算离散值是这样的。我们可以设置一个值bcv,edgeR来计算它的平方,这样就得到单样本的离散值了。edgeR建议,针对人类数据,建议设定为0.4。对于遗传上相似,建议设定为0.1。对于技术重复的实验,建议设定为0.01。
小果这里用的是人类数据,所以设置为0.4
dge_bcv <- dge
bcv <- 0.4 #
et <- exactTest(dge_bcv, dispersion = bcv ^ 2)
用decideTestsDGE上调的基因和下调的有多少. 并设置pvalue为0.05
gene1 <- decideTestsDGE(et, p.value = 0.05, lfc = 0)
summary(gene1)

差异基因怎么这么少,可能两组差异不明显,小果还是下调一下参数吧,改成0.2可能好一点。
bcv <- 0.2
et2 <- exactTest(dge_bcv, dispersion = bcv ^ 2)
gene2 <- decideTestsDGE(et2, p.value = 0.05, lfc = 0)
summary(gene2)

这时候差异基因显现出来了~
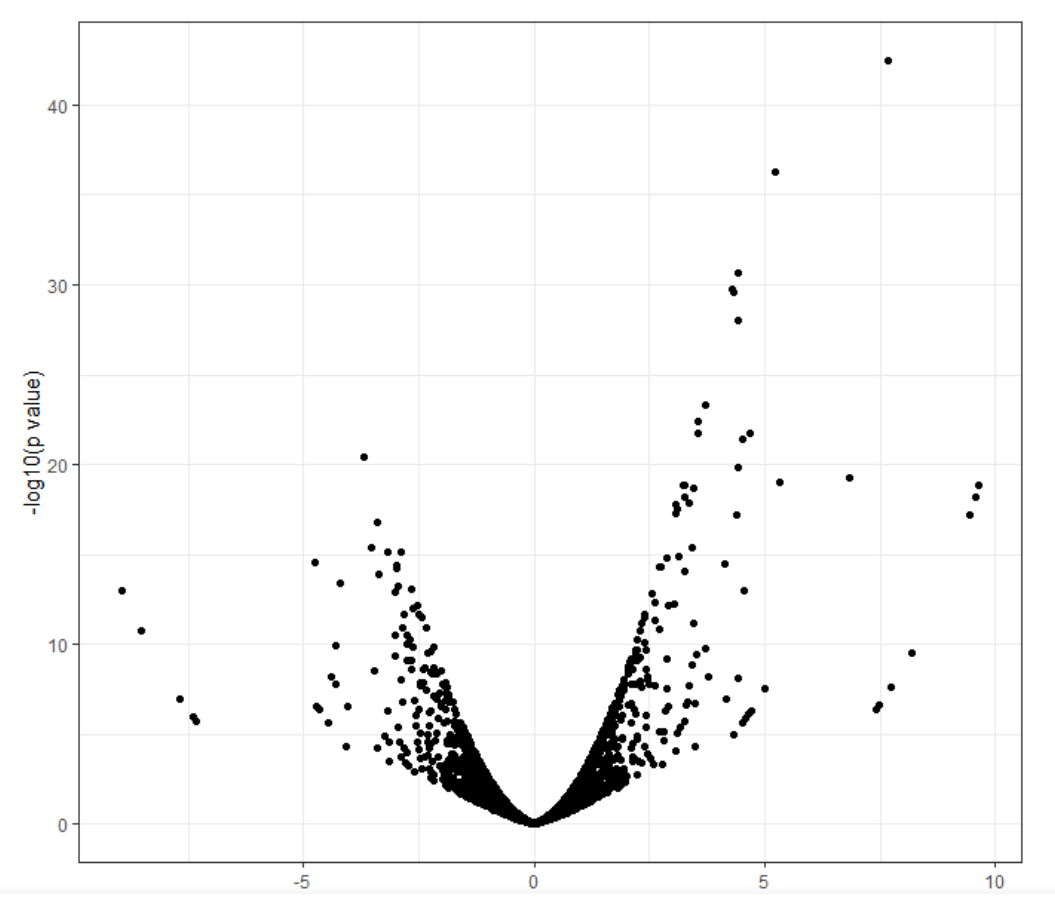
画个火山图看一下
library(ggplot2)
df <- et2$table
ggplot(df, aes(x=logFC, y=-log10(df$PValue)) ) +
geom_point() +
ylab(“-log10(p value)”) +
theme_bw()
和有重复差异表达进行比较
我们所使用的airway数据集是有多组重复的,所以我们可以很方便地对无重复与有重复进行比较,看会相差多少基因。
#与有重复比较
group <- meta$dex
y_rep <- DGEList(counts=expr, group = group)
#过滤及标准化
keep <- rowSums(cpm(y_rep)>1) >= 5
y_rep <- y_rep[keep, , keep.lib.sizes=FALSE]
y_rep <- calcNormFactors(y_rep)
#构建设计矩阵
design <- model.matrix(~group)
y_rep <- estimateDisp(y_rep, design = design)
fit <- glmQLFit(y_rep, design)
qlf.2vs1 <- glmQLFTest(fit, coef=2)
#查看差异基因数目
res <- decideTestsDGE(qlf.2vs1, p.value = 0.05, lfc = 0)
summary(res)
这下看下差异基因的数目,Down有1050,Up有869,明显多于以前。

将有重复的前100差异基因和无重复的前100差异基因进行比较
c1 <- row.names(topTags(et2,n = 100 ))
c2 <- row.names(topTags(qlf.2vs1, n = 100 ))
intersect_gene <- intersect(c1,c2
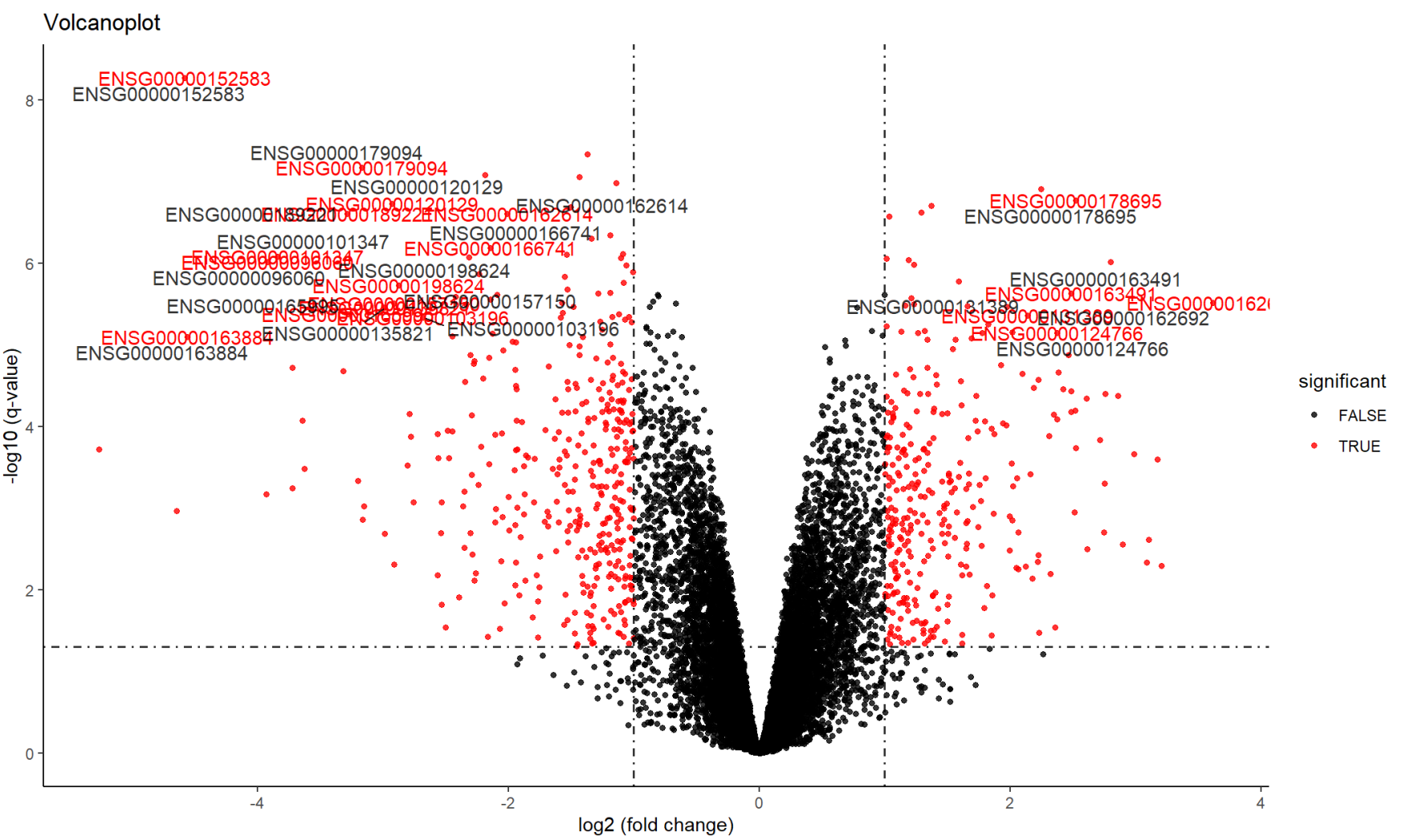
在有重复差异表达的火山图上面标记交集基因
library(ggplot2)
library(ggrepel)
data <- qlf.2vs1$table
data$significant <- as.factor(data$PValue<0.05 & abs(data$logFC) > 1)
data2 <- data[intersect_gene,]
data2$geneID <- rownames(data2)
p <- ggplot(data=data, aes(x=logFC, y =-log10(PValue),color=significant)) +
geom_point(alpha=0.8, size=1.2)+
scale_color_manual(values =c(“black”,”red”))+
labs(title=”Volcanoplot”, x=”log2 (fold change)”,y=”-log10 (q-value)”)+
theme(plot.title = element_text(hjust = 0.4))+
geom_hline(yintercept = -log10(0.05),lty=4,lwd=0.6,alpha=0.8)+
geom_vline(xintercept = c(1,-1),lty=4,lwd=0.6,alpha=0.8)+
#theme(legend.position=’none’)
theme_bw()+
theme(panel.border = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
axis.line = element_line(colour = “black”)) +
geom_text(data=data2, aes(label=geneID),col=”red”,alpha = 1) +
geom_text_repel(data=data2, aes(label=geneID),col=”black”,alpha = 0.8)
print(p)
这样就能清晰地看到我们无重复的基因与有重复的交集,的确能找到有明显差异表达的基因。但是不怎么明显的但是又有影响的基因就会被筛选掉。
edgeR是一种适用于无重复样本的差异表达分析工具,可以用来快速、简单地进行预实验。下面是使用edgeR进行差异表达分析的步骤:
- 数据准备:将原始的表达矩阵导入到R中,并根据实验设计创建一个组名向量。
- 数据处理:使用edgeR的函数来对表达矩阵进行数据处理,包括数据标准化、过滤低表达基因等。可以使用函数like.EList、calcNormFactors、cpm等。
- 差异表达分析:使用edgeR中的函数glmQLFit和glmQLFTest来进行差异表达分析,其中glmQLFit用于拟合广义线性模型,glmQLFTest用于计算差异表达的统计显著性。
- 调整bcv值:使用edgeR中的函数exactTest来对差异表达结果进行调整。
- 结果可视化:使用edgeR中的函数plotSmear、plotMDS等来进行差异表达结果的可视化。
最后小果提醒一下,由于无重复样本的不确定性,差异表达结果需要谨慎解释。虽然edgeR可以在无重复样本情况下进行差异表达分析,但是重复样本仍然是更可靠的选择,因为它可以降低误差,提高结果的可靠性。
你是否学会了呢,欢迎和小果讨论~
小果的云生信平台还有更多方便实用的工具,快来瞧瞧吧
云生信平台链接:http://www.biocloudservice.com/home.html
总结
无重复样本同样也可以用edgeR进行差异表达分析,可以简单地进行预实验。
- 通过调整bcv值:实现无重复样本的差异表达分析,不过bcv值需要根据经验进行调整。通常来说,人类基因0.4,对于遗传上相似设定为0.1。对于技术重复的实验,建议设定为0.01。
- 最后小果提醒一下,由于无重复样本的不确定性,差异表达结果需要谨慎解释。虽然edgeR可以在无重复样本情况下进行差异表达分析,但是重复样本仍然是更可靠的选择,因为它可以降低误差,提高结果的可靠性。
你是否学会了呢,欢迎和小果讨论~
小果的云生信平台还有DESeq2等多种差异表达分析的工具嗷,同时还有很多方便实用的工具~