在进行基因的差异分析之后,我们需要对基因的功能进行注释。基因功能注释的目的是通过分析基因的序列和结构以及已知的功能和特征信息,来预测和解释基因的功能和生物学作用。功能注释的结果可以提供关于基因的功能、参与的生物过程、细胞定位、调控机制等方面的信息,从而帮助研究者理解基因在生物体中的功能和相关的生理过程。
而对于基因功能的注释,小果想要寻求一种高效简便的方式,经过搜查资料发现,有一种无需代码的在线基因功能注释网站,它就是eggnog-mapper。下面就和小果来学习一下怎么使用吧!
一、eggNOG-Mapper介绍
eggNOG-Mapper是一种常用的基因功能注释工具,它基于eggNOG数据库和OrthoDB数据库,并利用这些数据库中的数据和比对算法来推断基因的功能。
其中,eggNOG数据库是一个用于进化分类和基因功能注释的数据库,它将已知的基因组按照进化关系进行分类,并给每个分类分配一个evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG)编号,同时还提供了关于这些基因的功能注释信息。
而OrthoDB数据库则是另一个用于基因功能注释的数据库,它主要用于识别和注释正交基因组。
二、如何使用eggNOG-Mapper?
首先我们需要打开eggNOG-mapper的主页,即http://eggnog-mapper.embl.de/,如图所示为该网页的信息。
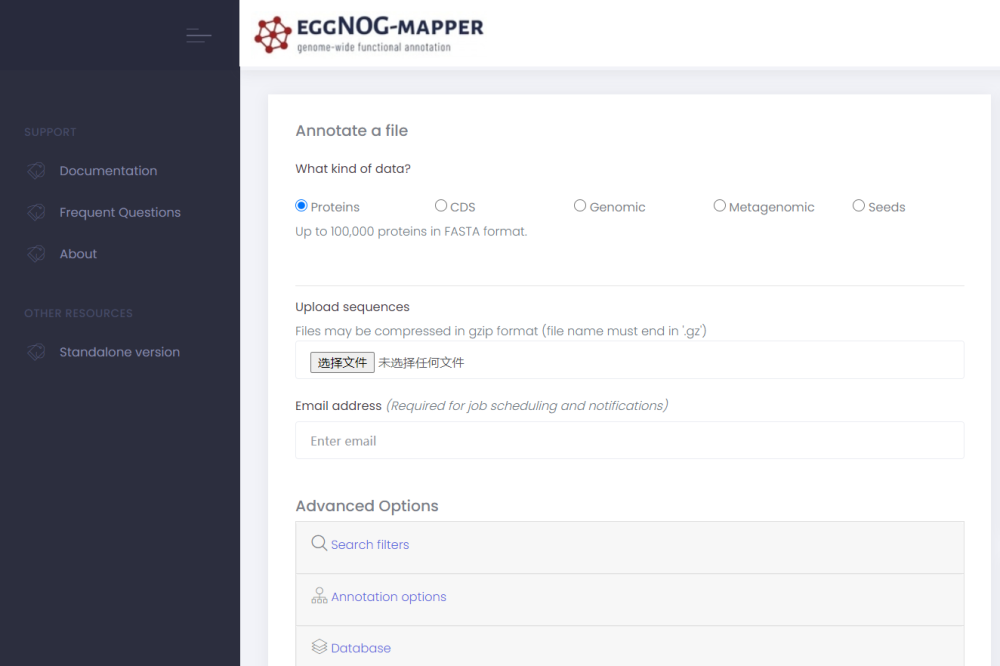
首先第一步,我们会选择数据的类型;在这里,小果选用的是在NCBI下载的穿心莲的蛋白质序列,然后放入步骤二所示的选择文件框中;最后第三步,则是我们需要输入接收邮件的email地址(一定要确保该邮箱能够接受邮件哦!)
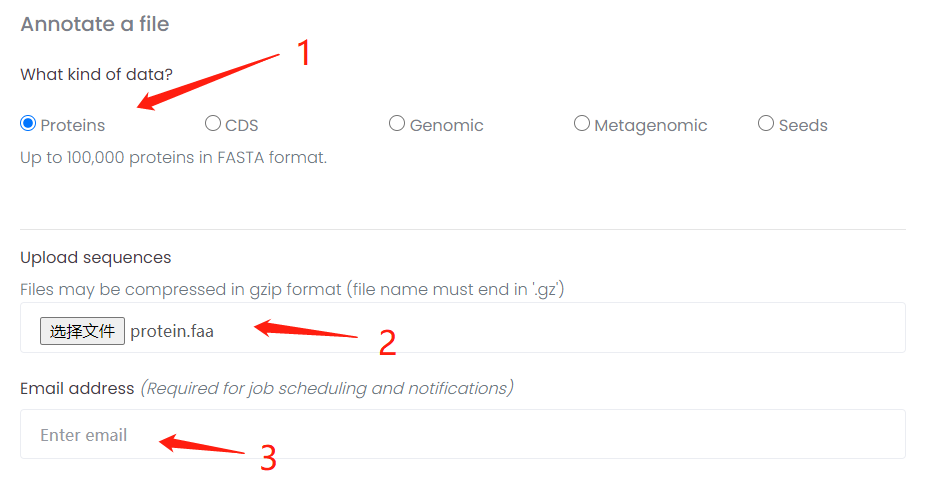
在经过上面的步骤之后,就可以点击submint按钮进行提交。之后我们可以打开接受的邮件,打开邮件之后,我们可以看到的信息如下。在这时候,我们需要点击1中的Click to manage your job去进行管理提交的任务。否则打开2中的链接我们会发现,就会没有基因功能注释文件的存在。

之后,我们就可以点击start job的按钮,启动任务了,在这里需要等待的时间大概要十来分钟。如何判断是否完成工作了。这个时候,我们可以等待下一封邮件的到来。
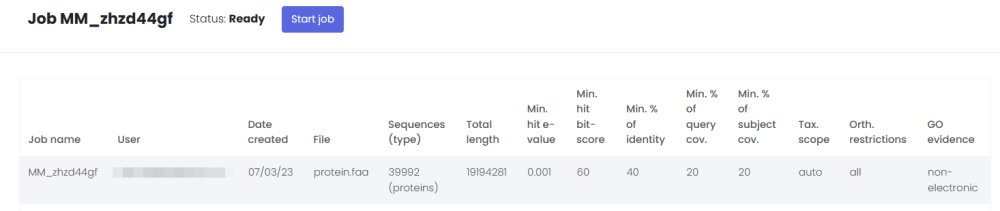
如果已经完成比对,并生成注释文件,这时候会收到一封邮件,上面会注明”finished”、”with status finished”等字样。打开邮件。并且点击上面2中的Access your job files here按钮,可以得到out.emapper.annotations文件并下载就行了
接下来,打开我们下载的out.emapper.annotations文件,就可以得到相应的基因功能注释信息了啦!
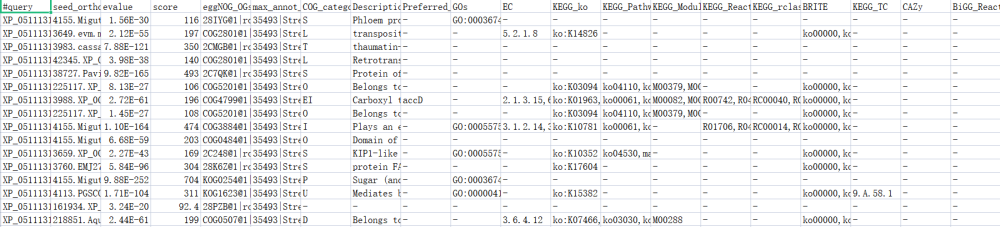
其中以下是out.emapper.annotations文件每个列的说明:
1)query:输入序列的名称。
2)seed_ortholog:匹配到的种子序列的注释信息。
3)evalue:输入序列与种子序列的匹配E-value阈值。
4)score:输入序列与种子序列的匹配得分。
5)eggNOG_OGs:与输入序列匹配的EggNOG orthologous groups(OGs)。
6)max_annot_lvl:在输入序列的注释中提供的最大注释级别。
7)COG_category:Clusters of Orthologous Groups(COG)分类。
8)Description:该序列的功能描述。
9)Preferred_name:该序列的首选或标准名称。
10)GOs:Gene Ontology(GO)注释信息。
11)EC:对应的酶学注释号。
12)KEGG_ko:对应的KEGG Orthology(KO)号。
13)KEGG_Pathway:KEGG通路信息。
14)KEGG_Module:KEGG模块信息。
15)KEGG_Reaction:KEGG反应信息。
16)KEGG_rclass:KEGG反应分类信息。
17)BRITE:BRITE功能层次结构注释。
18)KEGG_TC:KEGG传输物质分类信息。
19)CAZy:碳水化合物活性酶家族注释信息。
20)BiGG_Reaction:输入序列与BiGG数据库中的反应匹配的信息。
21)PFAMs:序列中与PFAM数据库匹配的信息。
好啦!经过一番学习,是不是感觉到eggNOG-Mapper的简便性了呢!真的就是零代码就能得到基因功能注释文件了呢!那么接下来大家也可以找到自己感兴趣的蛋白等去进行实操哦!对了,悄悄告诉你,如果对生信工具感兴趣的话,可以点击http://www.biocloudservice.com/home.html进行学习哦!