hello,今天小果教大家如何使用DESeq2包对counts表达矩阵进行差异基因分析,并对分析结果绘制火山图实现差异基因分析的可视化,如果你感兴趣的话就和小果一起看下去吧!
首先小果先来简单介绍一下这个包:
了解DESeq2
DESeq2是一个R语言中常用的差异表达分析包,用于分析高通量测序数据中的差异表达基因。它是Bioconductor项目的一部分,提供了一套统计方法和功能,用于检测基因在不同条件下的表达差异,并找出具有生物学意义的差异表达基因。DESeq2包的设计目标是针对RNA-seq数据进行差异表达分析,并且能够处理具有低复制数、高变异性和有偏差的数据。它采用了负二项分布模型和广义线性模型(GLM)的方法,通过考虑样本之间的技术和生物学变异,进行准确的差异表达分析。
DESeq2包提供了一系列功能,包括:
- 数据导入:支持从不同的数据源导入RNA-seq数据,包括原始计数数据和已经进行了基因注释的数据。
- 数据预处理:包括数据规范化、去除低表达基因、去除批次效应等。
- 差异表达分析:使用负二项分布模型和GLM进行统计分析,计算差异表达基因的显著性。
- 多组实验设计:支持多组条件下的差异表达分析,包括两组比较、多组比较和时间序列分析。
- 基因注释和功能富集分析:支持将差异表达基因进行注释,并进行功能富集分析,以了解差异表达基因的生物学功能。
- 可视化:DESeq2包集成了ggplot2和其他可视化工具,可以方便地绘制差异表达基因的图表,如火山图、MA图等。
差异基因分析流程
R包与数据加载
首先,设置工作目录为数据文件所在的路径,并加载DESeq2和其他所需的R包,并导入我们准备好的counts表达矩阵:
setwd(“/media/desk16/iyun007/kegg”)
#BiocManager::install(“DESeq2”)
library(DESeq2)
data1 <- read.csv(“Gene Symbol_matrix.csv”,header = T, row.names = 1)
dim(data1) #59427 5 (COUNT矩阵有59427 个基因、5个样本)
注意,我们导入的数据基本格式如下,同学们要提供符合要求的数据集哦!
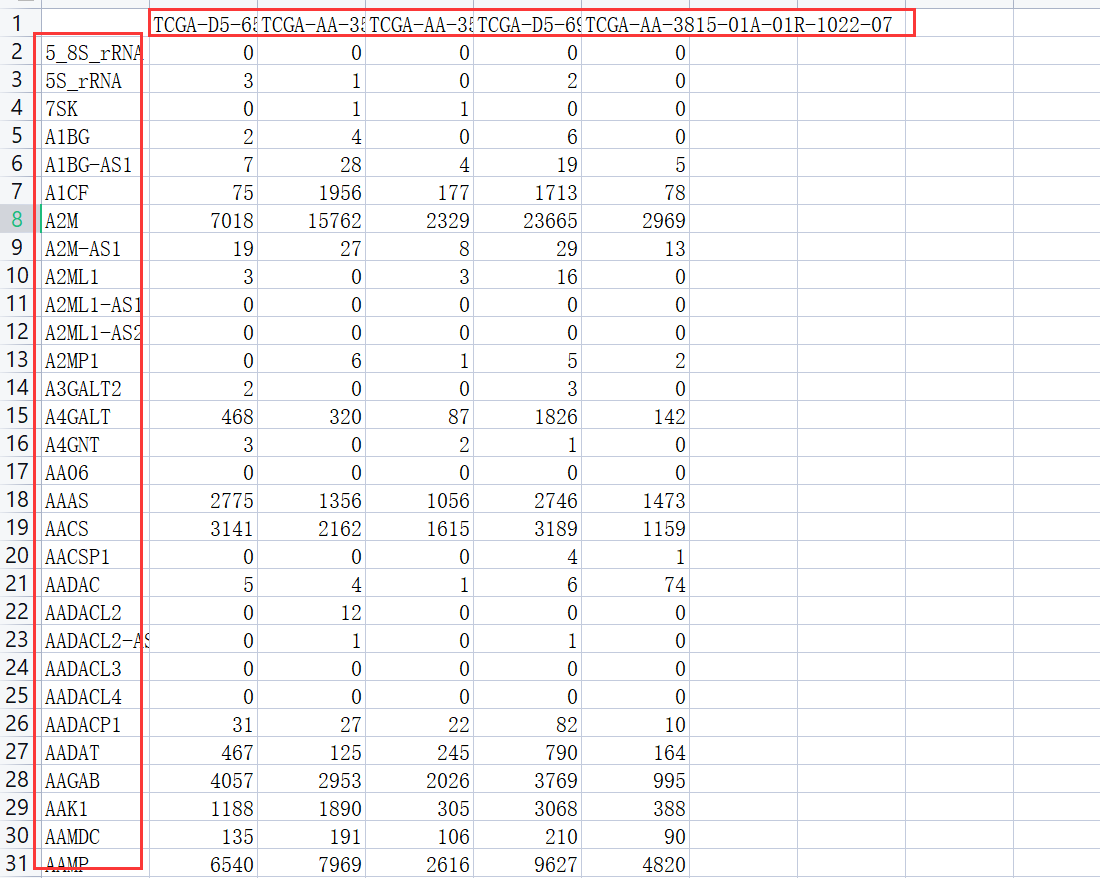
数据预处理
接下来,我们要构建分组矩阵,即根据数据框中样本的命名规则,提取出癌症样本和正常样本的列名,并将它们分别存储在tumor和normal向量中:
#构建分组矩阵
tumor <- colnames(data1)[as.integer(substr(colnames(data1),14,15)) < 10] #data1表达矩阵中的所有癌症样本(8个)
tumor
normal <- colnames(data1)[as.integer(substr(colnames(data1),14,15)) >= 10] #data1表达矩阵中的所有正常样本(2个)
normal
tumor_sample <- data1[,tumor] #癌症样本的表达矩阵
View(tumor_sample)
tumor_sample
ncol(tumor_sample)
normal_sample <- data1[,normal] #正常样本的表达矩阵
View(normal_sample)
ncol(normal_sample)
data1 <- cbind(tumor_sample,normal_sample) #合并癌症和正常样本矩阵
col_index <- which(colnames(matrix) == “normal_sample”)
colnames(matrix)[col_index] <- normal
#colnames(data1)[5] <- “TCGA.AA.3525.11A.01R.A32Z.07”
View(data1)
group_list <- c(rep(‘tumor’,ncol(tumor_sample)),rep(‘normal’,1)) #根据自己的数据集设置tumor和normal数据的分组数
condition = factor(group_list)
coldata <- data.frame(row.names = colnames(data1), condition) #coldata是分组矩阵
View(coldata)
最后合并好的数据如下:
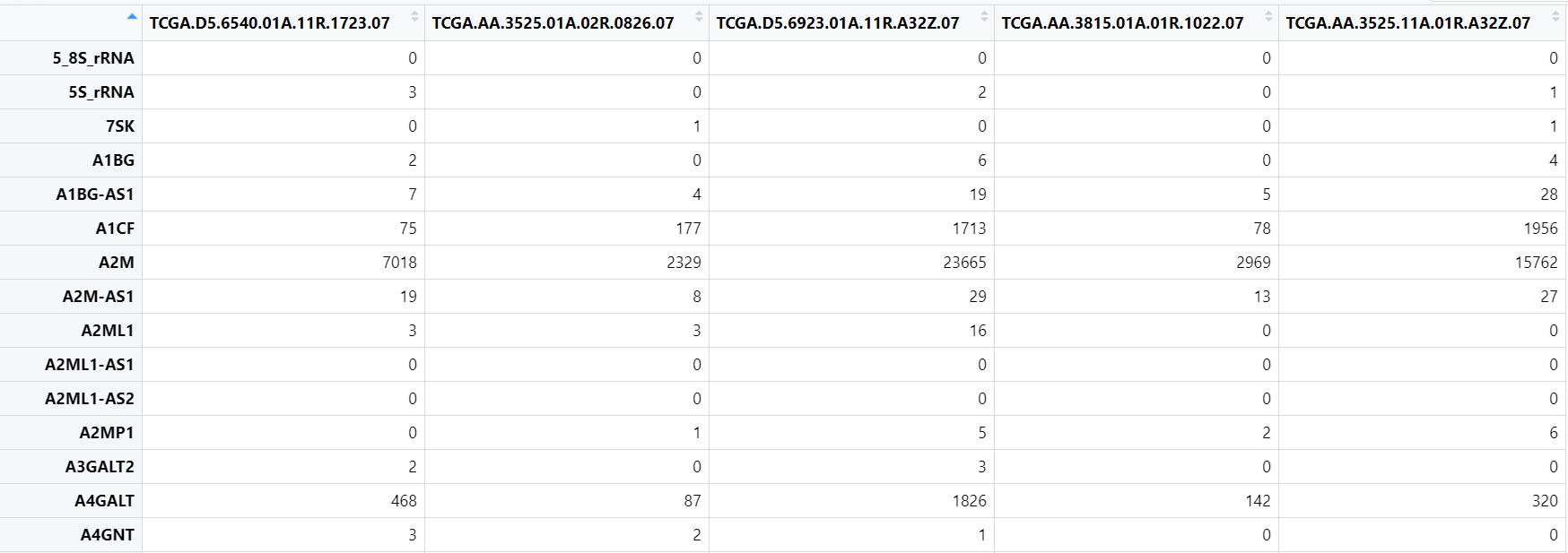
分组信息如下:
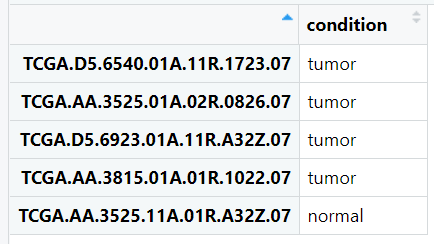
差异分析
接下来,我们可以:
- 创建DESeq2数据对象:使用DESeqDataSetFromMatrix()函数创建DESeq2所需的数据对象,将合并后的数据框和分组矩阵作为参数传递给该函数。
- 进行差异分析:使用DESeq()函数对DESeq2数据对象进行差异分析。
- 保存差异表达基因结果:将差异表达基因结果保存为CSV文件。
#构建dds对象,构建差异基因分析所需的数据格式
dds <- DESeqDataSetFromMatrix(countData = data1, colData = coldata, design = ~ condition)
#差异分析
dds <- DESeq(dds)
#提取结果
result <- as.data.frame(results(dds)) # results()从DESeq分析中提取出结果表
DGE <-subset(result,padj < 0.05 & (log2FoldChange > 2 | log2FoldChange < -2)) #这里我们提取出2倍差异表达、校正p值<0.05的显著差异表达基因
DGE <- DGE[order(DGE$log2FoldChange),]
write.csv(DGE,’DGE.csv’,row.names = TRUE)
我们一起来看下分析后的结果把!

绘制火山图
接下来,我们可以通过火山图可视化我们的分析结果,具体操作如下:
这里小果使用ggplot()函数创建绘图对象,并设置数据源为result数据框。然后使用geom_point()函数绘制散点图,其中x轴为log2FoldChange,y轴为-log10(padj),并根据差异表达的方向和显著性进行颜色分类。通过调整绘图的各种参数,如标题、坐标轴标签和图例,可以使火山图更具可读性和美观性哦。
## 5.提取结果
result <- as.data.frame(results(dds)) # results()从DESeq分析中提取出结果表
result <- na.omit(result)
result1 <- result[c(2,6)]
## 6.火山图
result1$change = ifelse(result1$padj < 0.05 & abs(result1$log2FoldChange) >= 2, ifelse(result1$log2FoldChange> 2 ,’Up’,’Down’),’Stable’)
library(‘ggplot2′)
ggplot(result1, aes(x = log2FoldChange, y = -log10(padj),colour=change)) +
geom_point(alpha=0.3, size=3.5) +
scale_color_manual(values=c(“#546de5″,”#d2dae2″,”#ff4757″))+
labs(title=’Volcano plot’,x=”log2(fold change)”,y=”-log10 (padj)”)+ # 坐标轴# 坐标轴和图标题title=”Volcano plot”,
theme_bw()+ #去除背景色
#xlim(-20, 20)+ #设置坐标轴范围
#ylim(0,40)+
# 辅助线
geom_vline(xintercept=c(-2,2),lty=3,col=”black”,lwd=0.8) +
geom_hline(yintercept = -log10(0.05),lty=3,col=”black”,lwd=0.8) +
theme(panel.grid = element_blank())+ #去除网格线
theme(plot.title = element_text(hjust = 0.5,size=24),
legend.position=”bottom”,
legend.title = element_blank(),
legend.text=element_text(size=18),
legend.key.size = unit(1, ‘cm’),
legend.background = element_rect(fill=”gray90″, linetype=”solid”,colour =”gray”),
axis.title.x =element_text(size=18),
axis.title.y=element_text(size=18),
axis.text=element_text(size=14,face = “bold”)
)
ggsave(“De_ana.png”,width=8,height=6)# 保存为png格式
我们一起来看下最后的结果图吧!
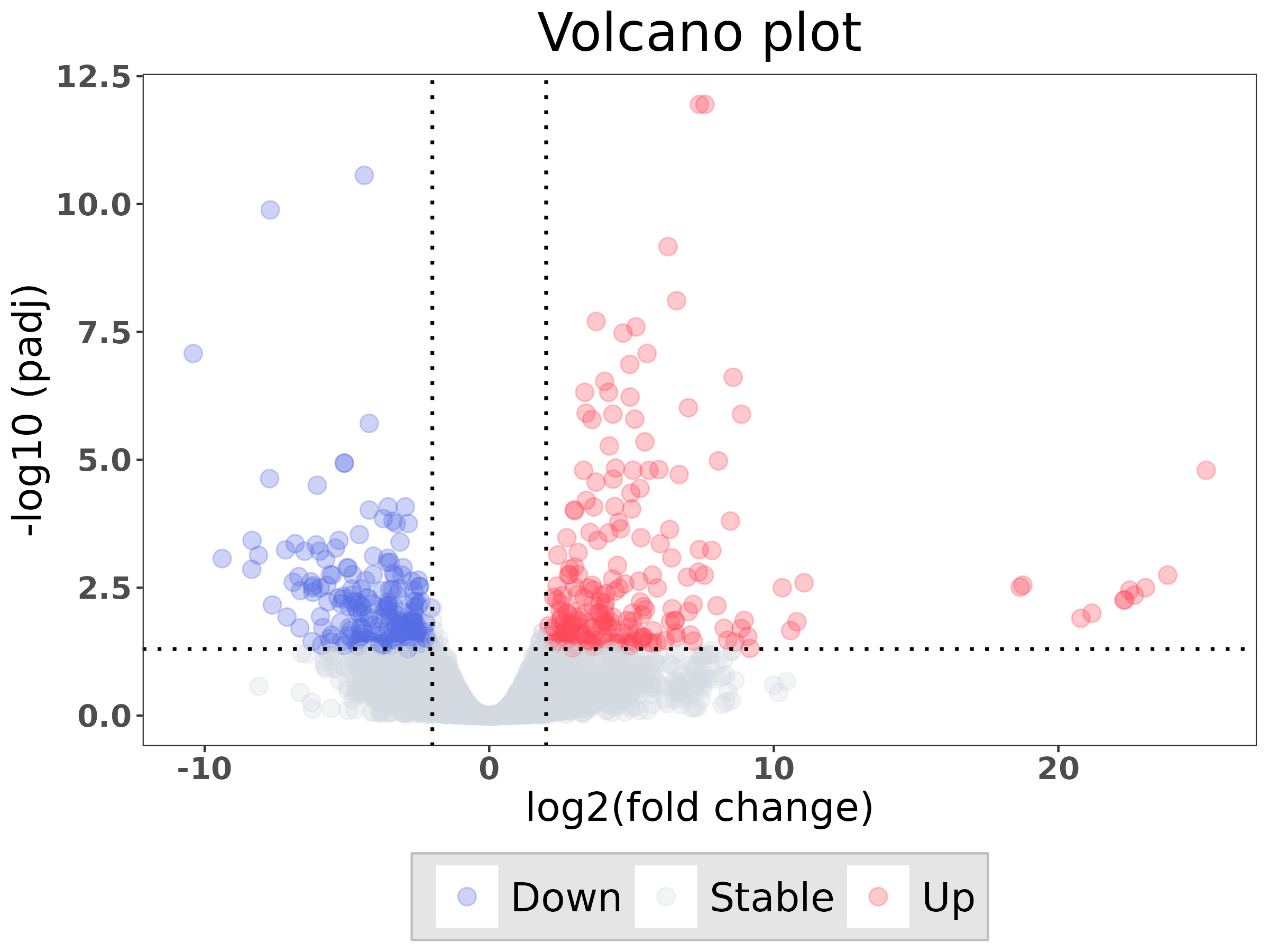
怎么样,你学会怎么使用DESeq2包来进行差异分析了嘛?是不是很简单!更多学习干活要继续关注小果哦!