在生命的生长发育过程中,细胞都在不断从一种状态过渡到另一种状态,表达不同的基因,产生蛋白组和代谢物的动态重复。我们可以使用轨迹推断的方法,根据测序细胞瞬时状态之间表达模式的相似性对单细胞沿着轨迹进行排序,以模拟细胞动态变化的过程。
今天要介绍的RNA 速率分析(RNA velocity)基于真实的转录动力学,可用于细胞基因表达的动态分化的研究。通过拟合前体(未剪接的)和成熟(剪接的)mRNA 丰度之间的比值来获得基因特异性速度,得出可能的细胞状态变化,从而追溯细胞的起源和潜在的命运,优点在于这个方法不需要指定起点和终点,可以对未知分化关系的细胞进行分析。
接下来就和小果一起深入了解一下这个方法吧!
RNA velocity所需要的包和之前介绍的SCENIC一样,也有python和R的版本,我们这里介绍R包velocyto.R的安装。
官方指路https://github.com/velocyto-team/velocyto.R
library(devtools)
install_github(“velocyto-team/velocyto.R”)
#注意,如果系统没有安装boost库,需要提前安装一下,例如:
sudo apt-get install libboost-dev
对于输入文件,有以下三种格式可选:
- 来自SMART-seq2的bam文件
- loom文件,可由cellranger跑出,或scanpy保存
- dropEst的matrices,这个方法用得比较少,这里就不细说啦。
我们这里选择2的方法,读取loom文件的格式~
ldat <-read.loom.matrices(“sample.loom”)

该文件包含spliced(不包含内含子),unspliced(包含内含子)和ambiguous(在分析中不会被使用)三个elements的list
#提取spliced与unspliced文件
emat <- ldat$spliced #外显子(spliced)表达矩阵
nmat <- ldat$unspliced #内含子(unspliced)表达矩阵
#提取Seurat对象的UMAP信息
load(“sce.RData”) #sce为待分析的Seurat对象
umap <- sce@reductions$umap@cell.embeddings
#估计细胞间距离
cell.dist <- as.dist(1-armaCor(t(sce@reductions$umap@cell.embeddings)))
fit.quantile <- 0.02
rvel.cd <- gene.relative.velocity.estimates(emat, nmat, deltaT=2, kCells=10,
cell.dist=cell.dist,fit.quantile=fit.quantile,n.cores=24)
#在UMAP图上绘制RNA velocity
pdf(“velocity.pdf”,height=6,width=8)
pic <- UMAPPlot(sce)
ggplot_build(pic)$data
colors <- as.list(ggplot_build(pic)$data[[1]]$colour)
names(colors) <- rownames(umap)
p<- show.velocity.on.embedding.cor(umap, rvel.cd, n=30, scale=’sqrt’,
cell.colors=ac(colors,alpha=0.5),cex=0.8,arrow.scale=2,
show.grid.flow=T,min.grid.cell.mass=1.0,grid.n=50,
arrow.lwd=1,do.par=F,
cell.border.alpha =0.1,USE_OPENMP=1,
n.cores=24,main=”Cell Velocity”)
dev.off()
借用官方教程中的图片,我们可以得到类似这样的结果
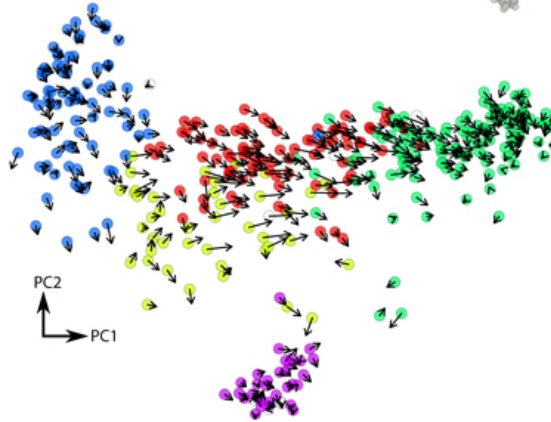
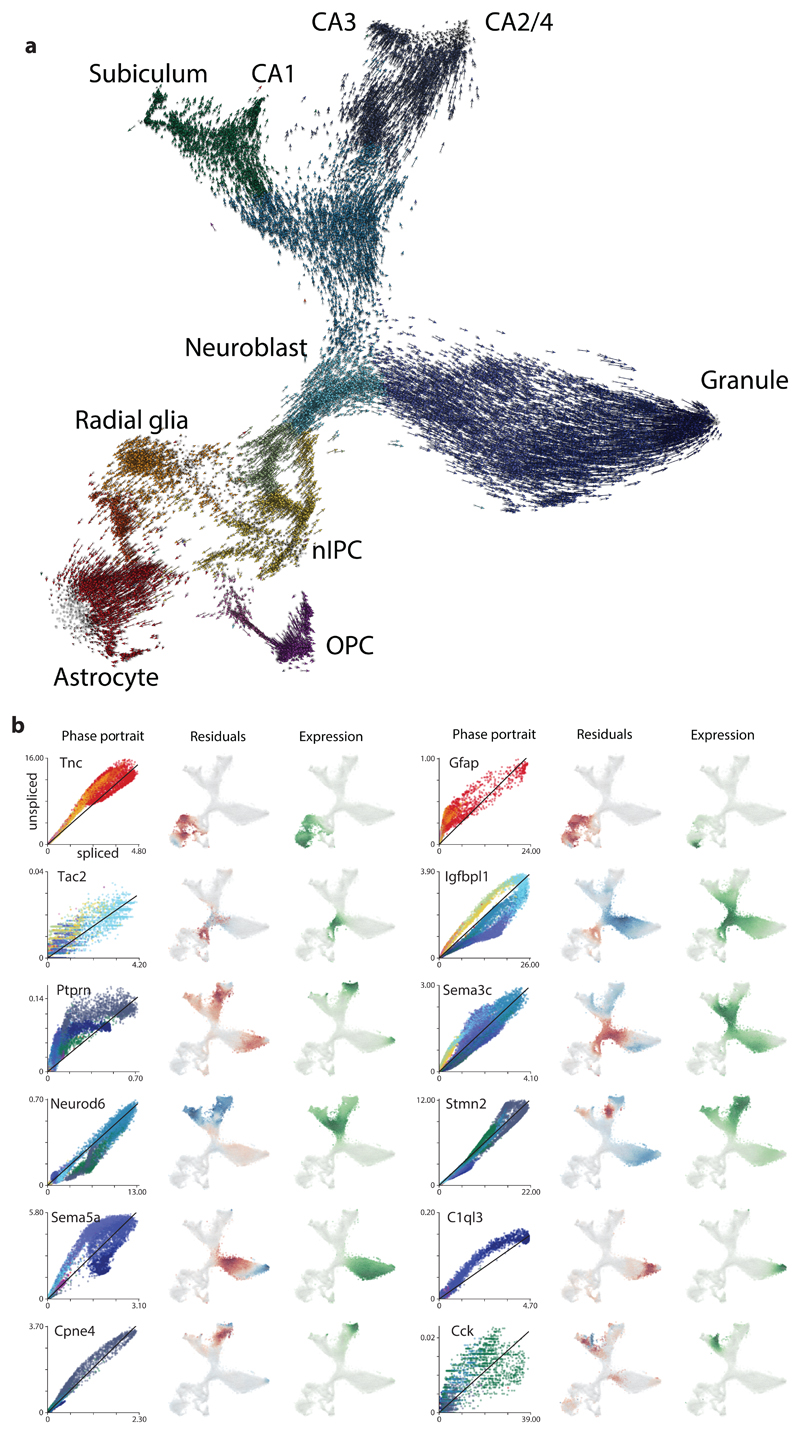
此外,如果使用已注释的单细胞转录组数据,我们可以知道各种类型的细胞分化是如何随时间进行的。如下图来源于Nature文章《RNA velocity of single cells》,可以得知颗粒细胞是由成神经细胞分化而来。
怎么样,是不是很神奇呢?快快用起来吧!那么今天的分享就到这里了~
欢迎使用:云生信-学生物信息学 (biocloudservice.com)
服务器使用可以联系微信:18502195490