今天小果带来的分享为首先利用ConsensusCluster对肿瘤样本进行分型,然后亚型间免疫检查点基因表达矩阵显著差异分析,该分析在肿瘤预后相关生信文章中出现的频率非常高,值得小伙伴们学习奥,接下来跟着小果开启今天的学习之旅吧!
- 如何进行亚型间免疫检查点基因表达显著差异分析?
在进行该分析之前,小果为小伙伴简单介绍一下需要注意的要点,第一点首先对肿瘤样本进行分型,第二点准备好免疫检查点基因,最后利用ggpubr包进行亚型间免疫检查点基因表达矩阵显著差异分析,筛选出在不同亚型间有显著差异的免疫检查点基因,这就是大概的分析步骤,下面和小果一起开始今天的实操吧!
2.准备需要的R包
#安装需要的R包
install.packages(“tidyverse”)
install.packages(“ggplot2”)
install.packages(“ggsci”)
BiocManager::install(“ConsensusClusterPlus”)
#加载需要的R包
library(reshape2)
library(ggpubr)
library(ggplot2)
library(tidyverse)
library(ggsci)
library(ConsensusClusterPlus)
3.ConsensusCluster一致性聚类分析
#筛选出的有预后的TEX基因表达矩阵文件,行名为基因名,列为样本名
expr1<- read.table(file = “expr_cluster.txt”,#工作目录下的文件名
header = T,#设置有列名
sep = “\t”,#分隔符
row.names = 1,#设置行名
check.names=F)#不检查行名
dir.create(‘ConsensusCluster/’)#构建文件夹
#ConsensusCluster一致性聚类
results = ConsensusClusterPlus(as.matrix(expr1),
maxK=6,
reps=100,
pItem=0.8,
pFeature=1,
title=’ConsensusCluster/’,
clusterAlg=”km”,
distance=”euclidean”,
seed=123456,
plot=”pdf”)
icl <- calcICL(results,title = ‘ConsensusCluster/’,plot = ‘pdf’)
Kvec = 2:6
x1 = 0.1; x2 = 0.6
PAC = rep(NA,length(Kvec))
names(PAC) = paste(“K=”,Kvec,sep=””)
for(i in Kvec){
M = results[[i]]$consensusMatrix
Fn = ecdf(M[lower.tri(M)])
PAC[i-1] = Fn(x2) – Fn(x1)
}
optK = Kvec[which.min(PAC)]
optK
PAC <- as.data.frame(PAC)
PAC$K <- 2:6
#绘制PAC验证曲线
ggplot(PAC,aes(factor(K),PAC,group=1))+
geom_line()+
theme_bw()+theme(panel.grid = element_blank())+
geom_point(size=4,shape=21,color=’darkred’,fill=’skyblue’)+
ylab(‘Proportion of ambiguous clustering’)+
xlab(‘Cluster number K’)
ggsave(“PAC.pdf”)
#最小值也就是最佳K,仍然为3.
#提取分型信息,k=3
clusterNum=3 #k值变化调整
cluster=results[[clusterNum]][[“consensusClass”]]
sub <- data.frame(Sample=names(cluster),Cluster=cluster)
sub$Cluster <- paste0(‘C’,sub$Cluster)
table(sub$Cluster)
write.table(sub,file = “K3.txt”,sep=”\t”,quote=F)#输出结果
4.亚型间免疫检查点基因表达矩阵差异分析
#EXP.txt,基因表达矩阵文件,行名为基因名,列名为样本信息。
exp<-read.table(“EXP.txt”,header=T,row.names=1,sep=”\t”,check.names=F)
#提取免疫检查点基因表达矩阵文件
df<-filter(exp,rownames(exp)%in% c(“LAG3″,”CD47″,”TIGIT”,”SIRPA”,”TNFRSF4″,”VTCN1″,”HAVCR2″,”ICOS”,”CTLA4″,”CD274″,”PDCD1″,”BTLA”,”TNFRSF9″))
#行列转置
df<-as.data.frame(t(df))
#将行名转化为列名
df<-rownames_to_column(df,var=”Sample”)
#修改基因名
df<-rename(df,c(PD1=PDCD1,TIM3=HAVCR2,MYD1=SIRPA))
##连接亚型分组信息和免疫检查点基因表达矩阵文件
immunegene<-left_join(df,sub,by=”Sample”)
#长宽数据转化
immunegene<-melt(immunegene,id.vars=c(“Sample”,”Cluster”))
#设置列名
colnames(immunegene)<-c(“Sample”,”Cluster”,”Gene”,”Expression”)
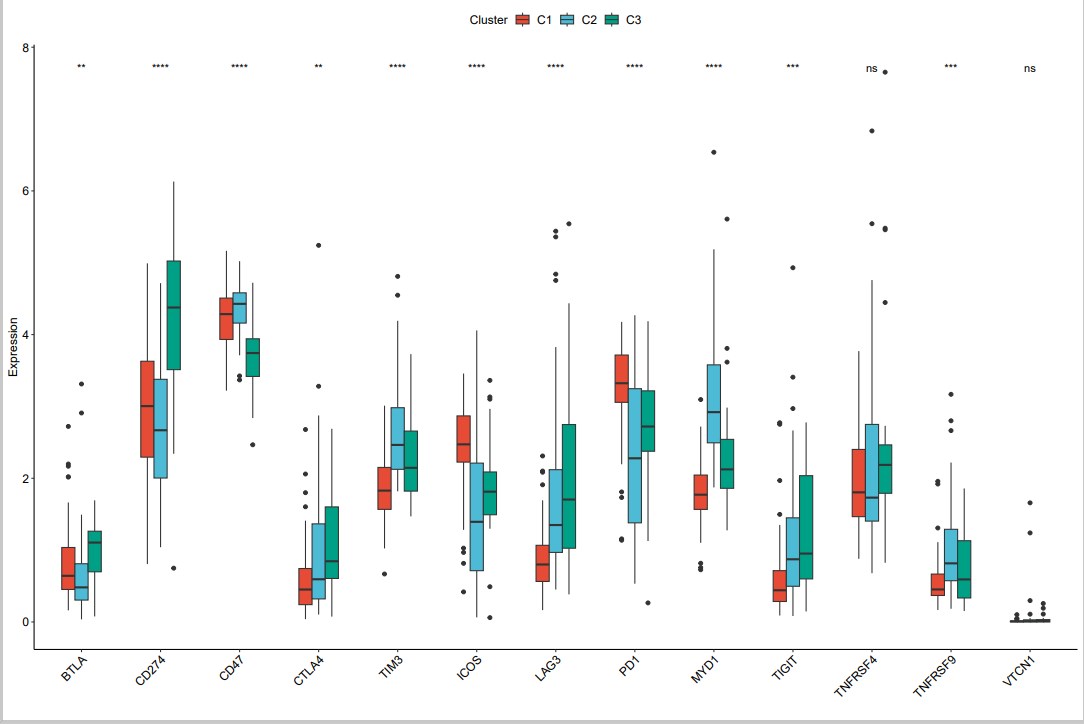
#绘制显著差异小提琴+箱线图
boxplot_immunegene<- ggplot(immunegene, aes(x = Gene, y =Expression ))+
labs(y=”Expression”,x= “”)+
geom_boxplot(aes(fill = Cluster),position=position_dodge(0.5),width=0.5)+
scale_fill_npg()+
#修改主题
theme_classic() +
theme(axis.title = element_text(size = 12,color =”black”),
axis.text = element_text(size= 12,color = “black”),
panel.grid.minor.y = element_blank(),
panel.grid.minor.x = element_blank(),
axis.text.x = element_text(angle = 45, hjust = 1 ),
panel.grid=element_blank(),
legend.position = “top”,
legend.text = element_text(size= 12),
legend.title= element_text(size= 12)
) +
stat_compare_means(aes(group = Cluster),
label = “p.signif”,#添加星号标签
method = “kruskal.test”,#kruskal.test多组检验
hide.ns = F)#显示不显著的
#保存图片
ggsave(file=”immunegene.pdf”,boxplot_immunegene,height=10,width=15)
- 结果文件
- immunegene.pdf
该结果图片为亚型间免疫检查点基因差异分析小提琴+箱线图,*表示p小于0.05,**表示p小于0.01,***表示p小于0.001,ns表示不显著。
2.K3.txt
该结果文件为利用ConsensusCluster一致性聚类分析,K=3时亚型分组信息文件,第一列为样本名,第二列为分组信息。

最终小果成功的完成了亚型间免疫检查点基因表达显著差异分析,看起来图片效果非常不错,欢迎大家和小果一起讨论学习呀!免疫浸润相关分析内容,例如单样本富集算法分析免疫浸润丰度(http://www.biocloudservice.com/106/106.php),计算64种免疫细胞相对含量(http://www.biocloudservice.com/107/107.php)等都可以用本公司新开发的零代码云平台生信分析小工具,一键完成该分析奥,感兴趣的小伙伴欢迎来尝试奥,网址:http://www.biocloudservice.com/home.html。今天小果的分享就到这里,下期在见奥。