
还在用Shell脚本跑生信流程吗?在为中断的Shell脚本搞得崩溃吗?修改日积月累的代码太头大?
那有没有什么办法,解决了上面的缺点,能让人少掉头发呢?
小果今天给大家推荐Snakemake,让你的生信分析流程, 像工厂的流水线一样开动起来!
Snakemake两个简单案例让你一看就懂。
Snakemake是什么?
Snakemake是一款基于Python3的软件,主要能够帮助用户快速搭建需要重复实现的分析流程。
以下是它的官方文档。
Snakemake — Snakemake 7.30.1 documentation
Snakemake的优势
- 通过treads可以设置任务的线程数目,实现并行处理
- 中断的程序,snake可以记录下来,从断掉的地方继续运行(拯救了多少生信人)
- 因为其是通过python3开发的,可以使用python库
- 支持shell命令操作
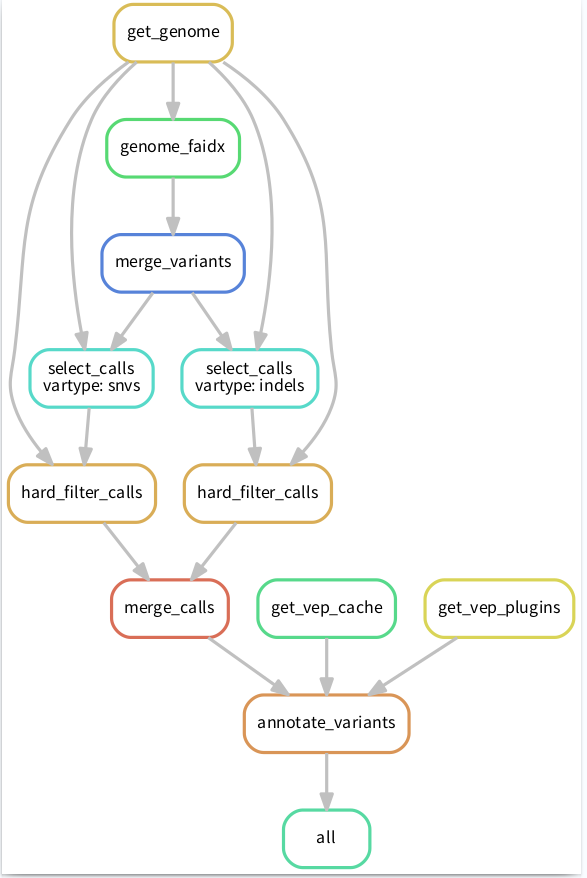
- 自动生成流程的拓扑图,使你的流程一目了然
例如像下面这张流程图一样
第一个案例:合并文件入门Snakemake
环境准备
推荐使用版本管理conda安装snakemake。(mamba是conda的C++版本其运行速度更快,下载更快,如果你还不知道,请快下载试试吧,一定让你爱上它。)
首先创建虚拟环境,为了流程可复现,指定python版本为3.5。
$ mamba create -n snake
$ conda activate snake
$ mamba install -y snakemake #使用mamba 安装snakemake
简单的snakemake实例
这里举一个简单易懂的合并文件内容的例子,帮助你快速入门snakemake
先创建一个文本文件,里面的内容为hello,再创建一个文本文件,内容为world。
$echo “hello” > hello.txt
$echo “world” > world.txt
接下来我们开始编写snakemake流程文件snakefile
rule concat:
input:
expand(“{file}.txt”, file=[“hello”, “world”])
output:
“merged.txt”
shell:
“cat {input} > {output}”
concat是为这个rule起的名字。
input为输入的文件。
output为输出的文件。
shell为执行的脚本命令。
{}类似于通配符,通过expand会通配file=[“hello”, “world”]文件。
在同一目录下运行snakemake
当执行工作流时,Snakemake会尝试生成给定的目标文件。
$ snakemake
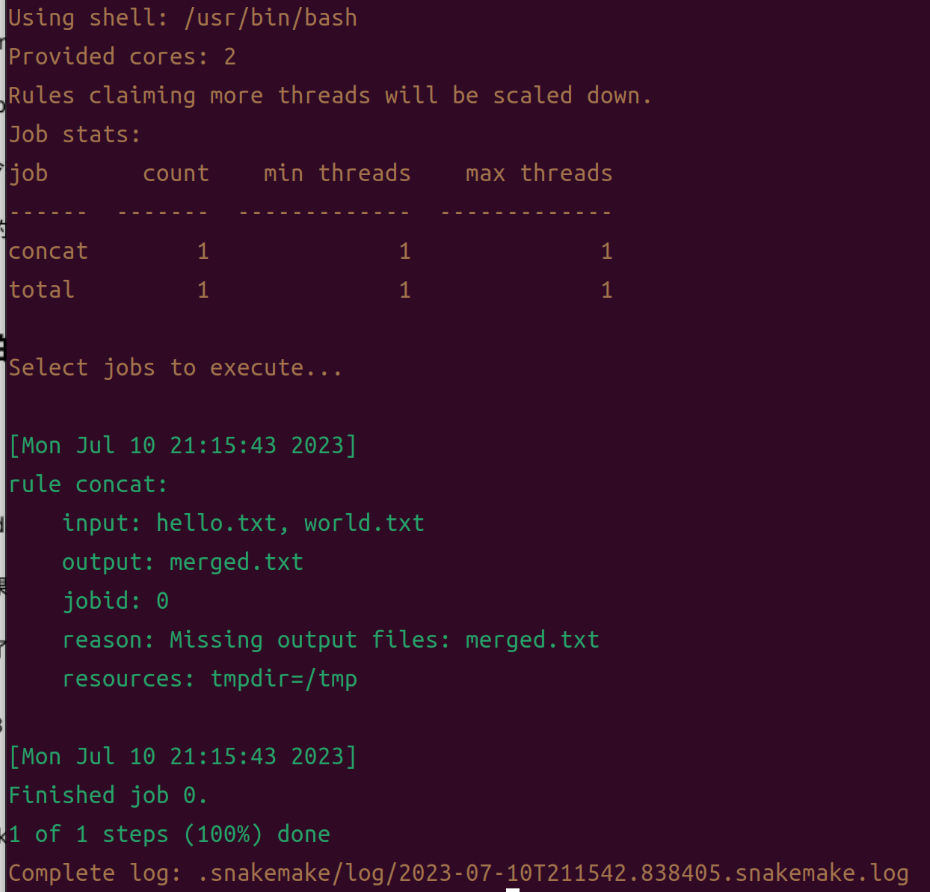
这样运行就完成了,我们打开输出文件看一下
$ less merge.txt
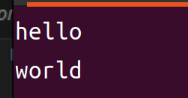
这样文件两个文件就合并成一个文件了。
通过上面一个实例,我们能够看到。snakemake流程的基本单元为rule, 在这个rule里分为三部分内容,第一个是input输入文件,第二个是output输出文件,第三个是sell,运行的shell命令(将shell改为run,它还可以运行python或R语言代码)。
snakemake的每一个rule就像工厂里的一台机器,我们通过不断的将这些机器串联在一起,就可以组装成一条生产的流水线了。
空运行
接下来介绍几个snakemake 常用参数:
–cores 为可供给所有任务的总cpu核心数。
-n/–dry-run 则为空运行,只检查所有的输入输出并报告需要执行的任务数。但并不真正地执行计算脚本。
-p/–printshellcmds 输出每一步的shell命令。
保存到Snakefile文件后,我们可以进行空运行(dry run)来测试流程,在每次跑流程前,都建议先执行dry-run来排查流程可能存在的问题。确认无误后在最后执行任务。
$ snakemake –cores 1 -np merge.txt
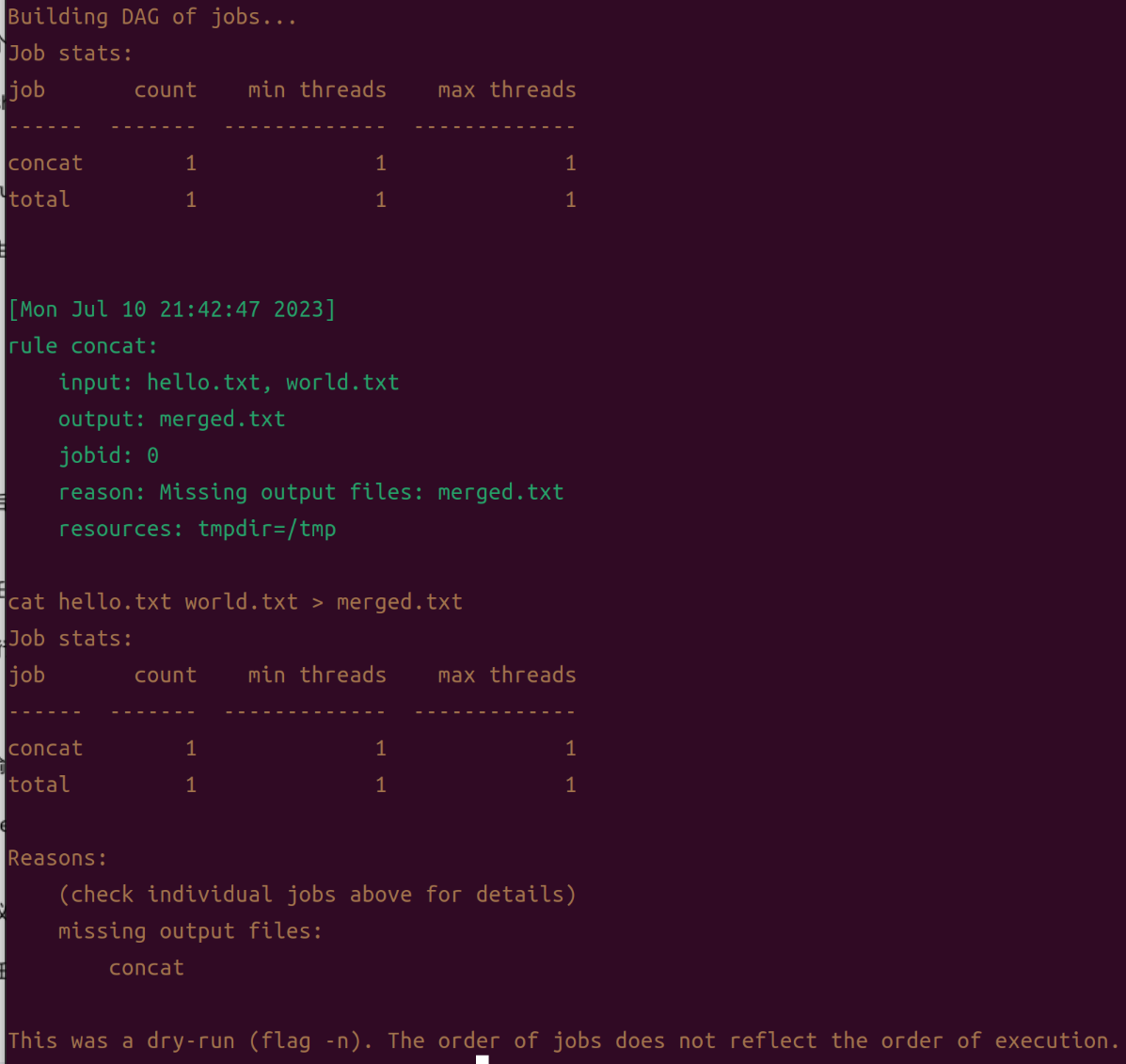
最后输入目标文件,snakemake会自动根据这些文件串起整个流程。
我们了解到有关snakemake最基础的概念,下面我们将做一个完整的工作流案例,来自snakemake的官方实例。
第二个案例:测序文件和参考基因比对,与差异分析
1.环境准备
$ mkdir snakemake-tutorial #创建一个实例目录
$ cd snakemake-tutorial
$ curl -L https://api.github.com/repos/snakemake/snakemake-tutorial-data/tarball -o snakemake-tutorial-data.tar.gz #下载来自官网的实例数据
$ tar –wildcards -xf snakemake-tutorial-data.tar.gz –strip 1 “*/data” “*/environment.yaml” #解压缩
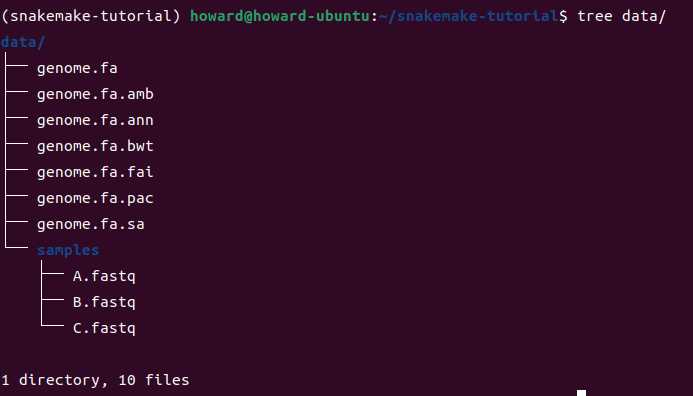
下载的数据包括这些文件
$ mamba env create –name snakemake-tutorial –file environment.yaml #创建虚拟环境命名为snakemake-tutorial, 并安装依赖environment.yaml
$ conda activate snakemake-tutorial #激活环境
打开snakemake帮助文档看一下,如果能返回结果,说明安装成功了
$ snakemake –help
创建一个Snakefile文件
$ vim Snakefile
2.创建比对规则
rule bwa_map:
input:
“data/genome.fa”,
“data/samples/{sample}.fastq”
output:
“mapped_reads/{sample}.bam”
shell:
“bwa mem {input} | samtools view -Sb – > {output}”
我们使用{sample}通配符,来匹配所有我们需要处理的文件。A,B,C.fastq
snakemake -np mapped_reads/{A,B}.bam
运行空命令
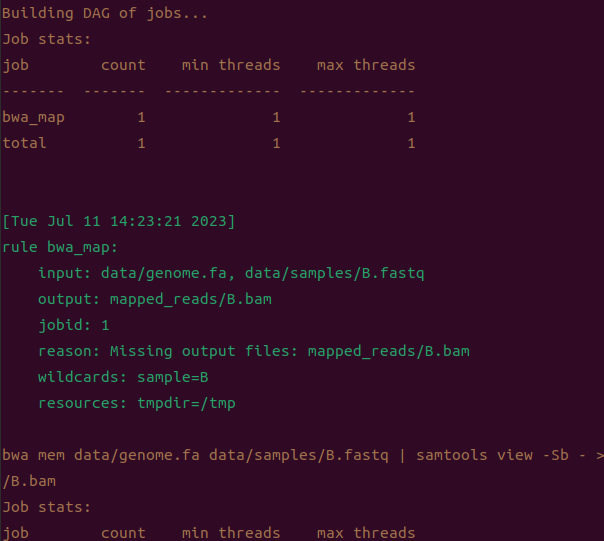
结果没问题 ,我们再继续编写我们的工作流程。
3.应用samtools对bam文件排序
rule samtools_sort:
input:
“mapped_reads/{sample}.bam”
output:
“sorted_reads/{sample}.bam”
shell:
“samtools sort -T sorted_reads/{wildcards.sample} ”
“-O bam {input} > {output}”
此规则将从mapped_reads目录中获取输入文件,并将排序后的版本存储在sorted_reads目录中。使用{wildcards.sample}来通配读取文件名
4.建立索引
rule samtools_index:
input:
“sorted_reads/{sample}.bam”
output:
“sorted_reads/{sample}.bam.bai”
shell:
“samtools index {input}”
我们已经定义了三个rule,可以导出拓扑流程图看一下。
snakemake –dag sorted_reads/{A,B}.bam.bai | dot -Tsvg > dag.svg
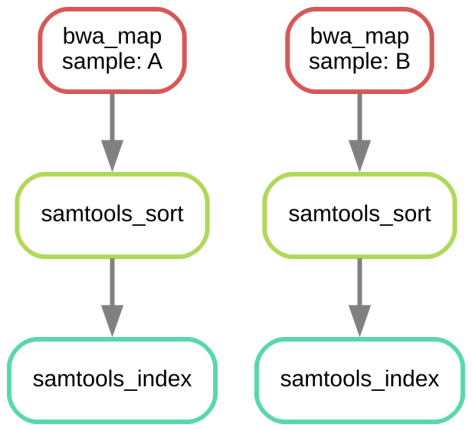
5.生成基因组变体
这里应用了上个案例的expand函数
SAMPLES = [“A”, “B”]
rule bcftools_call:
input:
fa=”data/genome.fa”,
bam=expand(“sorted_reads/{sample}.bam”, sample=SAMPLES),
bai=expand(“sorted_reads/{sample}.bam.bai”, sample=SAMPLES)
output:
“calls/all.vcf”
shell:
“bcftools mpileup -f {input.fa} {input.bam} | ”
“bcftools call -mv – > {output}”
这时我们再画一个图
snakemake –dag calls/all.vcf | dot -Tsvg > dag1.svg
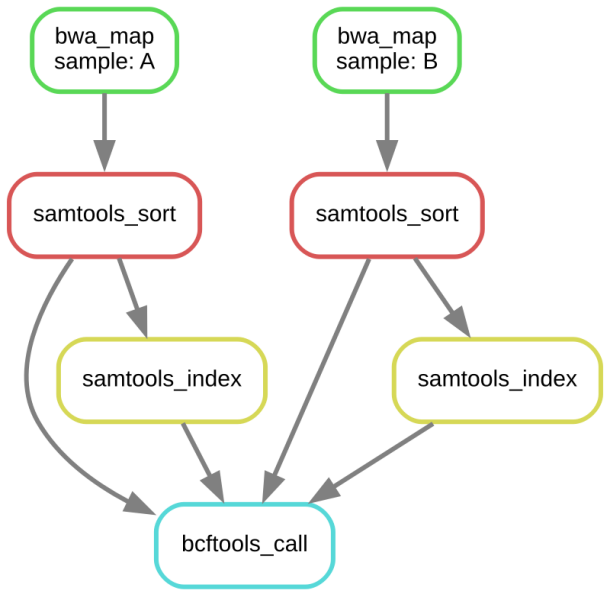
6.使用自己编写的python脚本
mkdir scripts
cd scripts
vim plot-quals.py
使用以下python脚本
import matplotlib
matplotlib.use(“Agg”)
import matplotlib.pyplot as pltfrom pysam import VariantFile
quals = [record.qual for record in VariantFile(snakemake.input[0])]plt.hist(quals)
plt.savefig(snakemake.output[0])
snakemake同样也支持R语言脚本
7.添加snakemake流程目标文件
rule all:
input:
“plots/quals.svg”
我们在命令行运行流程空文件
snakemake -n
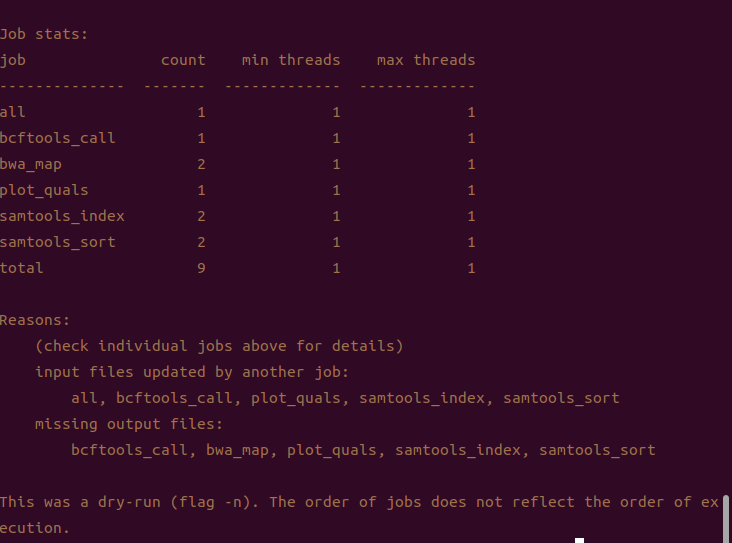
这样我们的流程就搭建好了,空命令运行没有问题,我们正式运行
snakemake -c1
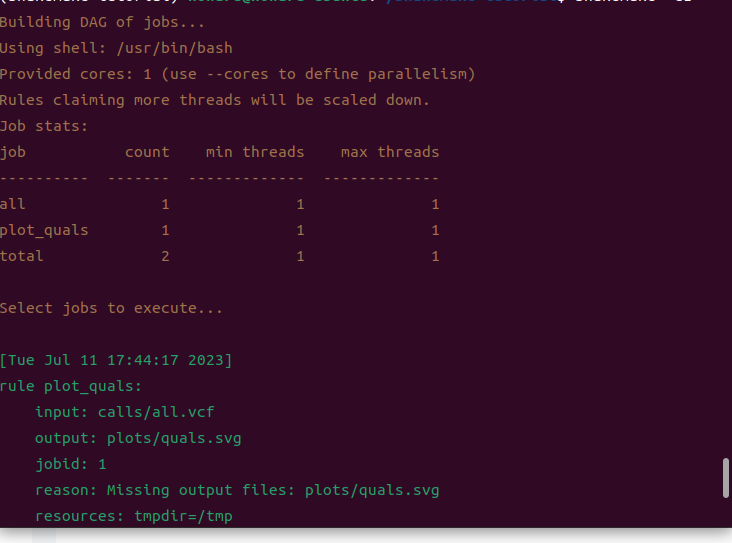
我们产生了所有的输出结果。
最终输出了结果图片,产生了突变的大部分在50bp以内。
好了今天小果的分享就到这里了。亲们是否明白了呢?欢迎大家和小果一起讨论学习哦!我们下期再见~