大家好,我是小果,一个对生物信息学充满热情的小伙伴。今天,我要和大家分享一项令人激动的技术——使用R语言中的神奇包randomForest进行疾病预测。通过这个强大的工具,我们可以利用数据的力量成为真正的“预测大师”。还等什么呢?快来探索这个神奇的领域吧!
疾病预测一直是医学领域的重大挑战,而现代生物信息学技术为我们提供了突破的机会。randomForest包的出现为我们带来了一种强大的工具,可以帮助我们预测疾病的发生和发展趋势。
randomForest是一种集成学习算法,基于决策树的思想。这个算法的奥妙之处在于,它通过组合多个决策树的预测结果来提高预测的准确性。就像森林中的树木一样,每棵决策树都有自己的特点,而集成后的结果却比单个树更可靠。
在R中,randomForest包提供了多个函数用于构建和分析随机森林模型。下面是randomForest包中常用的函数及其参数的解释:
- randomForest(): 这是构建随机森林模型的主要函数。它的主要参数包括:
x: 自变量矩阵或数据框,包含用于训练模型的特征。
y: 因变量向量,包含与自变量相对应的目标变量。
ntree: 决策树的数量,即随机森林中树的个数。
mtry: 每棵决策树中随机选择的特征数量。
importance: 是否计算变量重要性。
…: 其他参数用于控制模型构建过程,如节点分裂的准则、样本抽样方式等。
- importance(): 这个函数用于计算已构建随机森林模型中的变量重要性。
rf: 随机森林模型对象。
type: 重要性度量的类型,如MeanDecreaseAccuracy(平均准确率下降)、MeanDecreaseGini(平均Gini指数下降)等。
- predict(): 用于对新的数据进行预测或推断。
rf: 随机森林模型对象。
newdata: 包含要进行预测的新数据的矩阵或数据框。
- plot(): 用于绘制随机森林模型的图形,如变量重要性图、误差图等。
x: 随机森林模型对象。
…: 其他参数用于控制图形的外观和布局。
这些是randomForest包中的一些常用函数和参数。当使用这些函数构建和分析随机森林模型时,可以根据具体问题和需要来调整参数,以获得最佳的结果和理解模型的关键特征。
让我们来看一个使用randomForest包进行糖尿病预测的简单示例:
代码具体包括:
Step1 导入数据集以及数据预处理
# 这里的”inputdata.txt”是自行准备的本地文件,小果给大家附在最后。
# 1.导入数据集以及数据预处理
# 安装和加载randomForest包
#install.packages(“randomForest”)
library(randomForest)
# 导入数据集
data <- read.table(file = “D:/wanglab/life/ziyuan/20230628/inputdata.txt”,header = T) # 替换为实际数据集文件的路径
data$diabetes <- as.factor(data$diabetes)
head(data)
输入数据展示
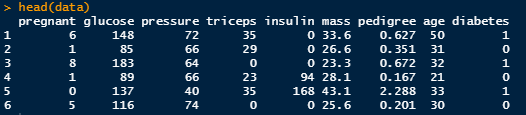
# 划分数据集为训练集和测试集
set.seed(123) # 设置随机种子,保证结果可复现
indices <- sample(1:nrow(data), nrow(data)*0.8) # 80%的数据用于训练,20%的数据用于测试
trainingset <- data[indices, ]
testingset <- data[-indices, ]
rf_model <- randomForest(diabetes ~ ., data = trainingset, ntree = 500,mtry=2)
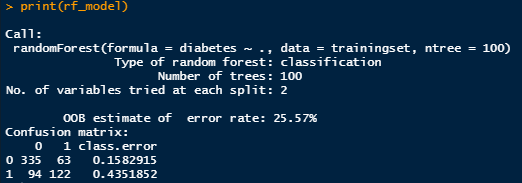
# 查看模型结果
print(rf_model)
# 变量重要性图
varImpPlot(rf_model)
rf_model$importance
变量重要性图
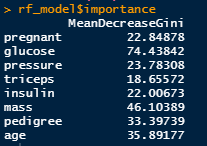
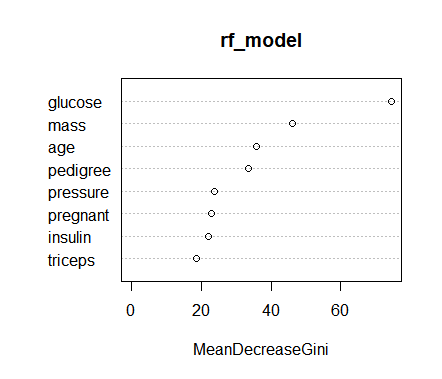
randomForest模型的结果将显示出重要的变量、每个变量的重要性指标以及预测准确性的度量。这些信息有助于我们理解哪些因素对于疾病的预测起着重要作用。
# 绘制误差图
plot(rf_model, main = “Random Forest Model Error”, type = “o”)
误差图
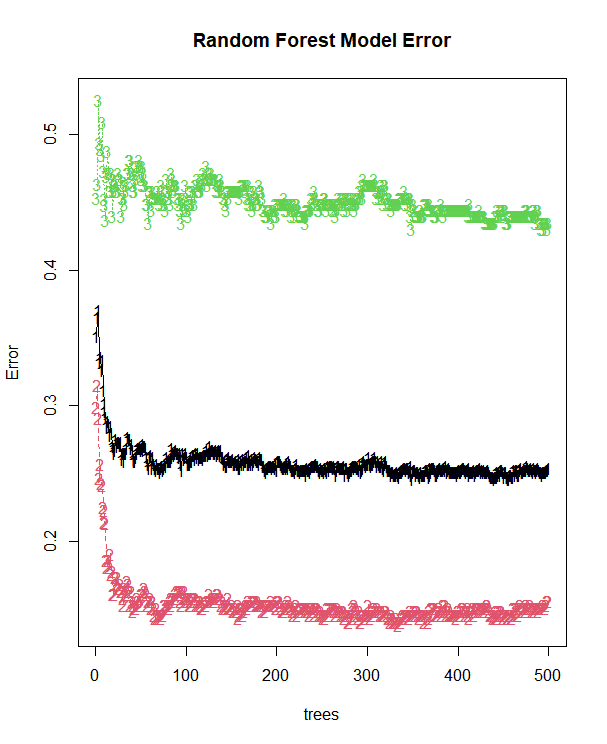
在误差图中,横轴表示随机森林中的每个决策树,纵轴表示误差或损失度量。每个点代表一个决策树模型的误差或损失值,而整体的线条或曲线表示所有决策树模型的平均误差或损失。误差图中的不同颜色线条代表了不同样本集生成的模型的预测误差。这些不同颜色的线条表示随机森林中的不同决策树模型或子模型的平均误差值。不同颜色的线条趋于平行且相互靠近,表示随机森林中的子模型具有相似的误差水平,模型的预测结果比较稳定。
# 绘制单棵决策树
library(rpart.plot)
plot(rf_model$forest[[1]], uniform = TRUE, main = “Decision Tree”)
单棵决策树
Step3 绘制AUC曲线评估模型
# 3.绘制AUC曲线评估模型
#对测试集进行预测
pre_ran <- predict(rf_model,newdata=testingset)
#将真实值和预测值整合到一起
obs_p_ran = data.frame(prob=pre_ran,obs=testingset$diabetes)
#输出混淆矩阵
table(testingset$diabetes,pre_ran,dnn=c(“真实值”,”预测值”))
#绘制ROC曲线
ran_roc <- roc(testingset$diabetes,as.numeric(pre_ran))
plot(ran_roc, print.auc=TRUE, auc.polygon=TRUE, grid=c(0.1, 0.2),grid.col=c(“green”, “red”), max.auc.polygon=TRUE,auc.polygon.col=”skyblue”, print.thres=TRUE,main=’随机森林模型ROC曲线,mtry=2,ntree=500′)
AUC曲线图
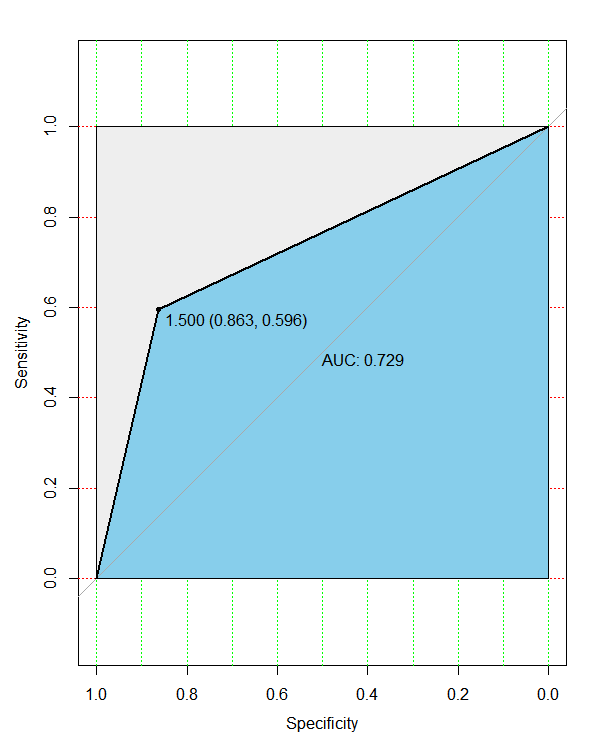
注:
# 请确保将路径”inputdata.txt”替换为实际数据集文件的路径,并根据需要调整模型参数和其他配置。
# 在绘制AUC曲线之前,确保您的模型输出的是预测的概率值,而不是类别标签。
在上述代码中,我们首先安装并加载了randomForest包。然后,我们创建了一个示例数据集,其中包含年龄(age)和体质指数(bmi)作为自变量,疾病情况(disease)作为因变量。接下来,我们使用randomForest函数构建了一个疾病预测模型,其中自变量通过~符号与因变量相连。最后,我们通过打印rf_model来查看预测结果。
现在,我想告诉大家一个有趣的事实:“使用randomForest包进行疾病预测,就像在森林中寻找秘密路径,而这个路径将带领我们揭开疾病的面纱。”是不是让你心动不已呢?
最后,我想强调一下使用公众号平台进行数据分析的便利性。云生信平台(http://www.biocloudservice.com/home.html)为我们提供了一个简单、方便、快速的方式来进行数据分析和分享结果。你只需轻松点击,即可掌握数据的奥秘,成为疾病预测的高手!
让我们一起利用randomForest包,借助公众号平台的力量,用数据驱动的方法来揭示疾病的秘密,为医学研究贡献一份力量吧!