在之前的文章中,小果向大家详细分享了质量评估的操作过程和结果分析,相信小伙伴们已经熟练掌握,迫不及待地进入下一阶段的学习啦。这一期小果给大家带来了序列比对的操作步骤,好啦,废话不多说,我们直接开始~
工具介绍
常见的序列比对软件类型:
- 数据库搜索:
BLAST, FASTA, PSI-BLAST……
- 两序列比对:
LASTZ, MUMmer……
- 多序列比对:
ClustalW, MUSCLE, T-Coffee
- 基因组比对:
GMAP, BLAT, ACT
- 短序列比对:
BWA, SOAP, Bowtie, ELAND, GSNAP, Stampy
小果这次采用短序列基因组比对工具BWA(Burrows-Wheeler Aligner)进行比对分析。以下是BWA软件主页:
http://bio-bwa.sourceforge.net/
BWA具有高效快速、高质量比对、支持多种比对模式、多线程支持、灵活性与可扩展性等优点,很适合初入生信大门的小伙伴们使用。
linux环境下,进行下列四步操作就可以使用啦:
# 1.下载软件安装包:
wget http://jaist.dl.sourceforge.net/project/bio-bwa/bwa-0.7.12.tar.bz2
# 2.解压缩:
tar jxf bwa-0.7.12.tar.bz2
# 3.进入解压缩目录:
cd bwa-0.7.12
# 4.编译安装:
make
# 注意:使用时写明全路径:/home/bin/bwa-0.7.12/bwa,或者设置环境变量(export PATH=”/home/bin/bwa-0.7.12:\$PATH”,也可写入\~/.bashrc并source)后仅使用程序名称。
BWA提供了三个主要的子命令:bwa index、bwa mem和bwa aln:
- bwa index:用于为参考基因组构建索引文件,以便于后续的比对操作。使用该命令需要指定参考基因组的文件,并生成一系列索引文件(如.bwt、.sa等),这些索引文件将在后续的比对操作中被使用。
- bwa mem:这是BWA中最常用的子命令,适用于处理 Illumina 测序数据。它可以将测序数据(fastq格式)与参考基因组进行比对。使用该命令需要指定参考基因组的索引文件、测序数据文件,并可以选择性地指定其他参数,如线程数、比对得分等。比对结果将输出为SAM格式文件。
- bwa aln:该命令适用于处理较短的测序片段,如 Illumina 测序中的短读长。它将测序数据与参考基因组进行比对,并生成比对位置的SA(Suffix Array)坐标。需要指定参考基因组的索引文件、测序数据文件,并可以选择性地指定其他参数,如mismatch惩罚、gap开销等。
除了上述核心命令外,BWA还提供了其他一些辅助命令和工具,用于索引的更新、格式转换和统计分析等。例如,bwa samse和bwa sampe用于处理短读长测序数据的比对结果;bwa fastq2fasta用于将fastq格式文件转换为fasta格式文件;bwa stats用于生成关于比对效果的统计信息等。想要了解更多的小伙伴们可以查阅官方文档哦~
另外这次还需要用到Samtools,Samtools 是一个用于处理SAM和BAM格式文件的工具集。它提供了一系列命令行工具,用于对比对结果进行操作、转换和分析。
以下是一些常用的Samtools命令:
- samtools view: 用于查看SAM/BAM文件的内容。可以选择性地输出指定区域的比对信息或过滤特定条件下的比对结果。
- samtools sort: 对BAM文件中的比对记录按照染色体位置进行排序,生成排序后的BAM文件。这有助于后续的索引和数据分析。
- samtools index: 为BAM文件创建索引文件,以便在后续的查询和访问中快速定位比对记录。
- samtools merge: 将多个BAM文件合并成一个单一的BAM文件,方便后续的比对结果整合和分析。
- samtools flagstat: 统计BAM文件中比对记录的各种标志(flag)的统计信息,例如总比对数量、倍数比对数量、未比对数量等。
- samtools mpileup: 在给定的位置上对比对记录进行堆叠,生成堆叠信息,用于变异检测和深度分析。
- samtools faidx: 为FASTA格式的参考基因组文件创建索引文件,以便在后续的查询和访问中快速提取序列。
- samtools calmd: 用于给BAM文件添加或更新比对记录的MD标签,这个标签表示了比对时可能的碱基差异。
以上只是Samtools命令集中的一部分,Samtools还提供了其他功能强大的命令和选项,用于处理和分析比对数据。它在生物信息学领域广泛应用于测序数据的处理、变异检测、比对评估以及其他相关任务。
操作步骤
书接上回,我们直接用前面已经设置好路径的数据继续开始,小果先把序列比对过程的全代码一次性放在这里,方便小伙伴们使用~:
# 为参考序列建立索引
bwa index ./00_ref/MGI358.SNP.fa
# 将测序reads比对到参考序列
bwa mem -M -Y -t 1 ./00_ref/MGI358.SNP.fa ./01_clean/test_clean.fq.gz > ./02_align/test.clean.sam
# 为节约存储,将sam转换为二进制文件bam,并对比对结果进行排序
samtools sort ./02_align/test.clean.sam -o ./02_align/test.clean.sort.bam
# 为排序后的bam文件创建索引
samtools index ./02_align/test.clean.sort.bam
# 使用samtools筛选出非重复比对的reads,输出为bam文件
samtools view -F 256 -hb ./02_align/test.clean.sort.bam > ./02_align/test.clean.sort.uniq.bam
# 统计覆盖深度
bamdst -p ./00_ref/target.358.SE50.subSNP.bed -o ./02_align ./02_align/test.clean.sort.uniq.bam
这些就是操作需要的全部代码啦,是不是很简单呢~如果小伙伴们平时在生信分析的操作过程中遇到困难,欢迎大家使用小果开发的生信工具平台http://www.biocloudservice.com/home.html,更加方便快捷哦~
下面,小果就给大家详细地对细节问题进行介绍,想提升自己的小伙伴们不要走开哦~
分步详细介绍
# 为参考序列建立索引
bwa index ./00_ref/MGI358.SNP.fa
索引的目的是为了加快后续的比对过程。BWA在比对时需要先加载索引,以获得更高的比对效率和准确性。
我们把索引完成后的文件夹下载到本地便于观看,会发现索引操作生成了多个文件,包括.bwt、 .fai、.pac、.ann、.amb和.sa等文件,它们作为BWA比对过程的输入之一。这些索引文件能够帮助BWA快速地从参考基因组中搜索匹配的片段,并进行比对,具体介绍如下:
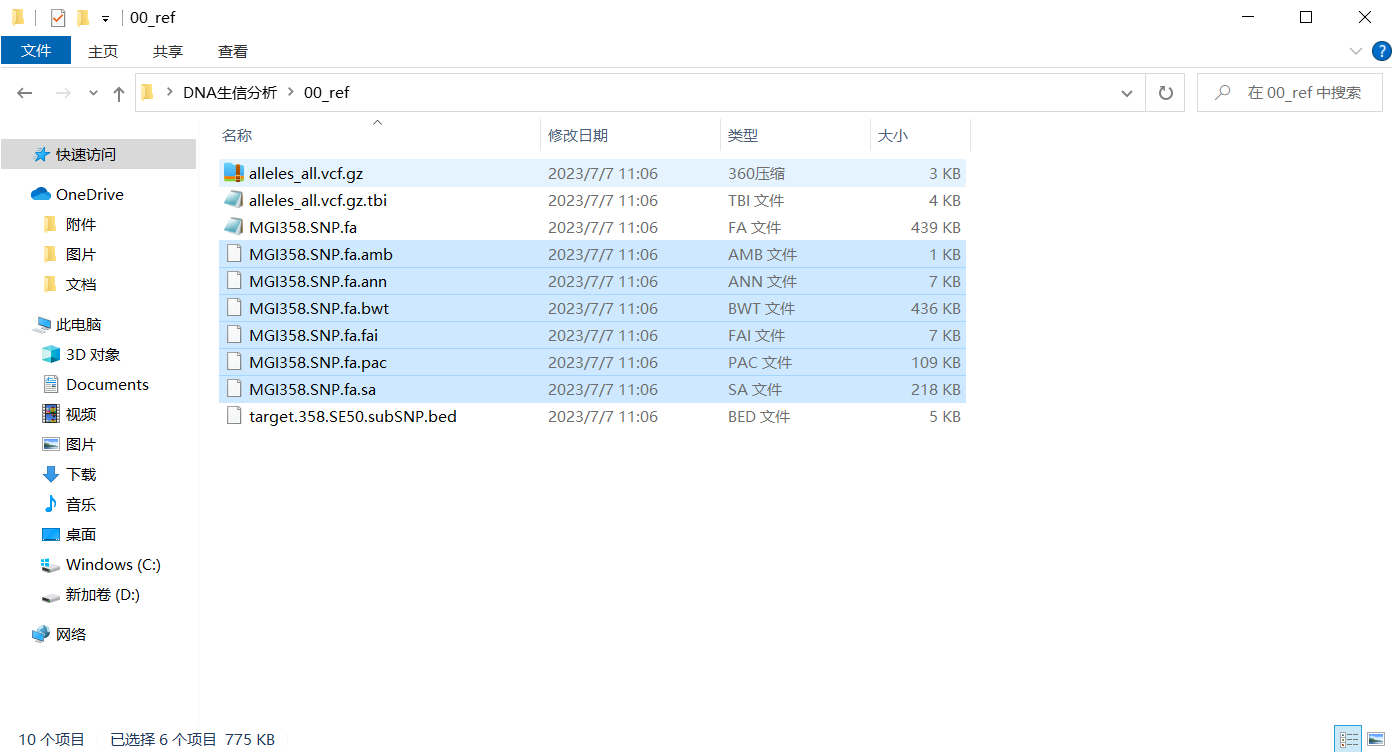
- .bwt 文件:Burrows-Wheeler转换后的索引文件,包含了对参考基因组的压缩和转换信息,用于快速搜索匹配的片段。
- .pac 文件:另一种压缩格式的索引文件,记录了参考基因组的碱基信息,提供给BWA进行比对。
- .ann 文件:注释文件,记录了参考基因组中的注释信息,例如区域名称、染色体位置等。
- .amb 文件:判断碱基模棱两可(ambiguous)的位点,即多个碱基可能性并存的位置。
- .sa 文件:后缀数组索引文件,包含了后缀数组的信息,用于搜索匹配的后缀。
- .fai 文件:记录了每条染色体/序列的名称、长度以及在索引过程中的顺序,某些变种调用程序需要读取 .fai 文件以确定染色体的位置和序列长度。
# 将测序reads比对到参考序列
bwa mem -M -Y -t 1 ./00_ref/MGI358.SNP.fa ./01_clean/test_clean.fq.gz > ./02_align/test.clean.sam
# 用less命令查看存储比对信息的序列文件sam
less ./02_align/test.clean.sam
这段代码看起来有一点长,小果在这里为大家拆开详细介绍:
- bwa mem: 表示调用BWA程序的mem子命令,用于执行比对操作。
- -M: 这个选项告诉BWA要生成一个包含Mate-SW信息的SAM文件。Mate-SW信息主要用于单端数据与配对的比对结果一起使用。
- -Y: 这个选项告诉BWA生成一个压缩的输出文件(即SAM文件)。压缩可以减少存储空间的使用。
- -t 1: 这个选项指定线程数为1,表示使用单线程运行BWA。如果你有更多的CPU核心可用,可以调整该值以加快速度。
- ./00_ref/MGI358.SNP.fa: 这是参考基因组的文件路径和文件名。
- ./01_clean/test_clean.fq.gz: 这是经过清洗后的测序数据(fastq格式)的文件路径和文件名。
- > ./02_align/test.clean.sam: 这部分代码将比对结果输出到一个名为 test.clean.sam 的SAM文件。> 符号将标准输出重定向到指定的文件路径。
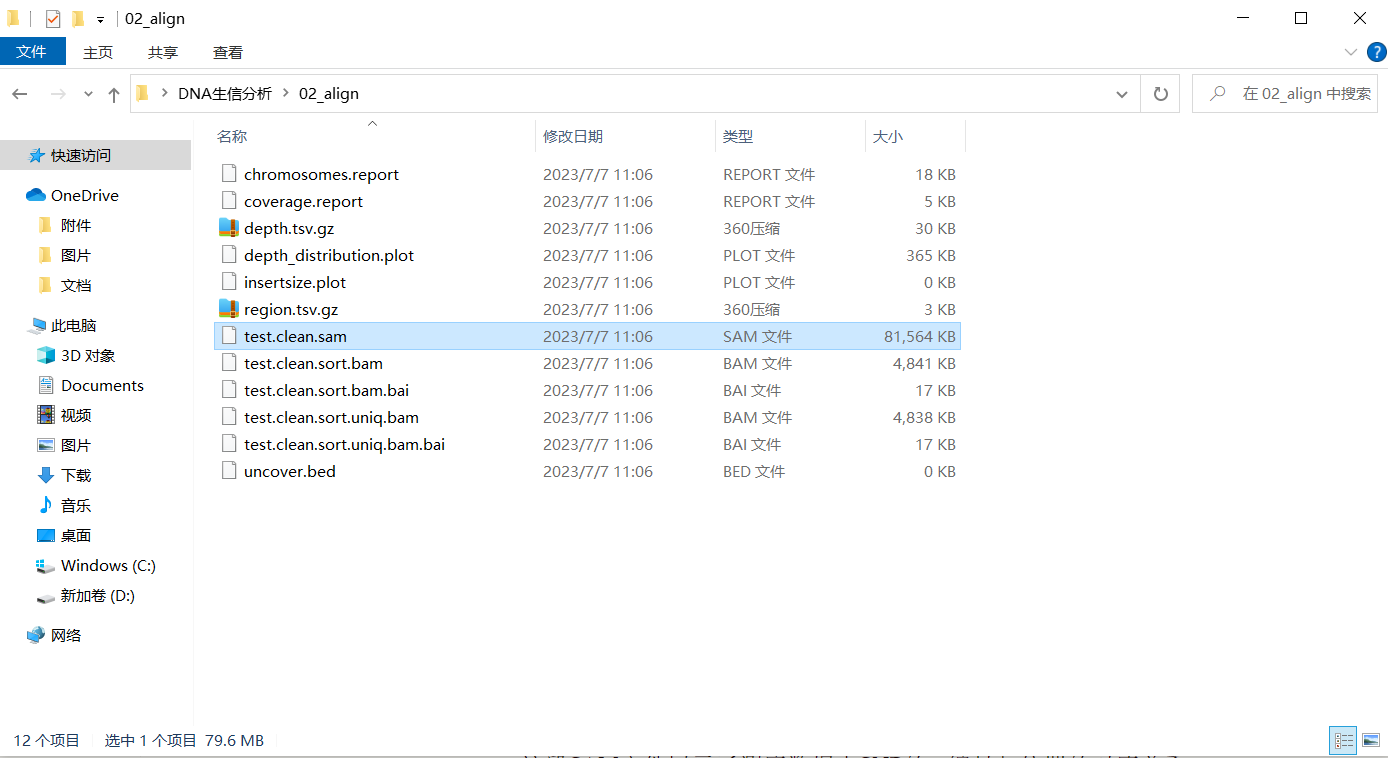
通过执行这段代码,BWA会使用单线程将清洗后的测序数据与参考基因组进行比对,生成一个SAM格式的比对结果文件。SAM文件中包含了每个测序读取的比对位置、比对质量等信息,可以用于后续的变异检测、比对统计和其他分析操作。
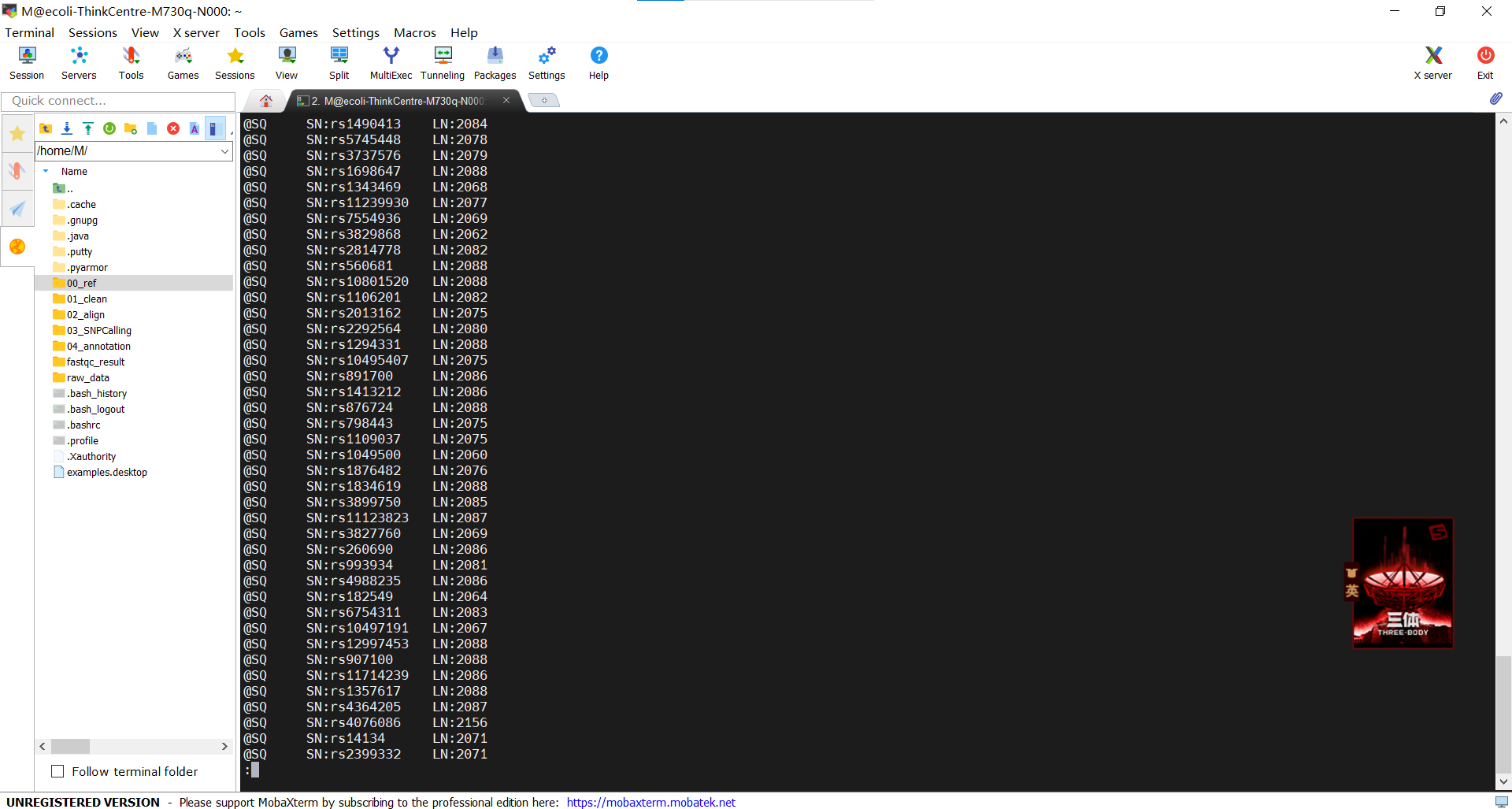
这段SAM文件展示了测序数据中SNP的rs编号与位置的对应关系。
# 为节约存储,将sam转换为二进制文件bam,并对比对结果进行排序
samtools sort ./02_align/test.clean.sam -o ./02_align/test.clean.sort.bam
# 为排序后的bam文件创建索引
samtools index ./02_align/test.clean.sort.bam
# 使用samtools筛选出非重复比对的reads,输出为bam文件
samtools view -F 256 -hb ./02_align/test.clean.sort.bam > ./02_align/test.clean.sort.uniq.bam
# 查看bam文件的内容
samtools view -S ./02_align/test.clean.sort.bam | less -S
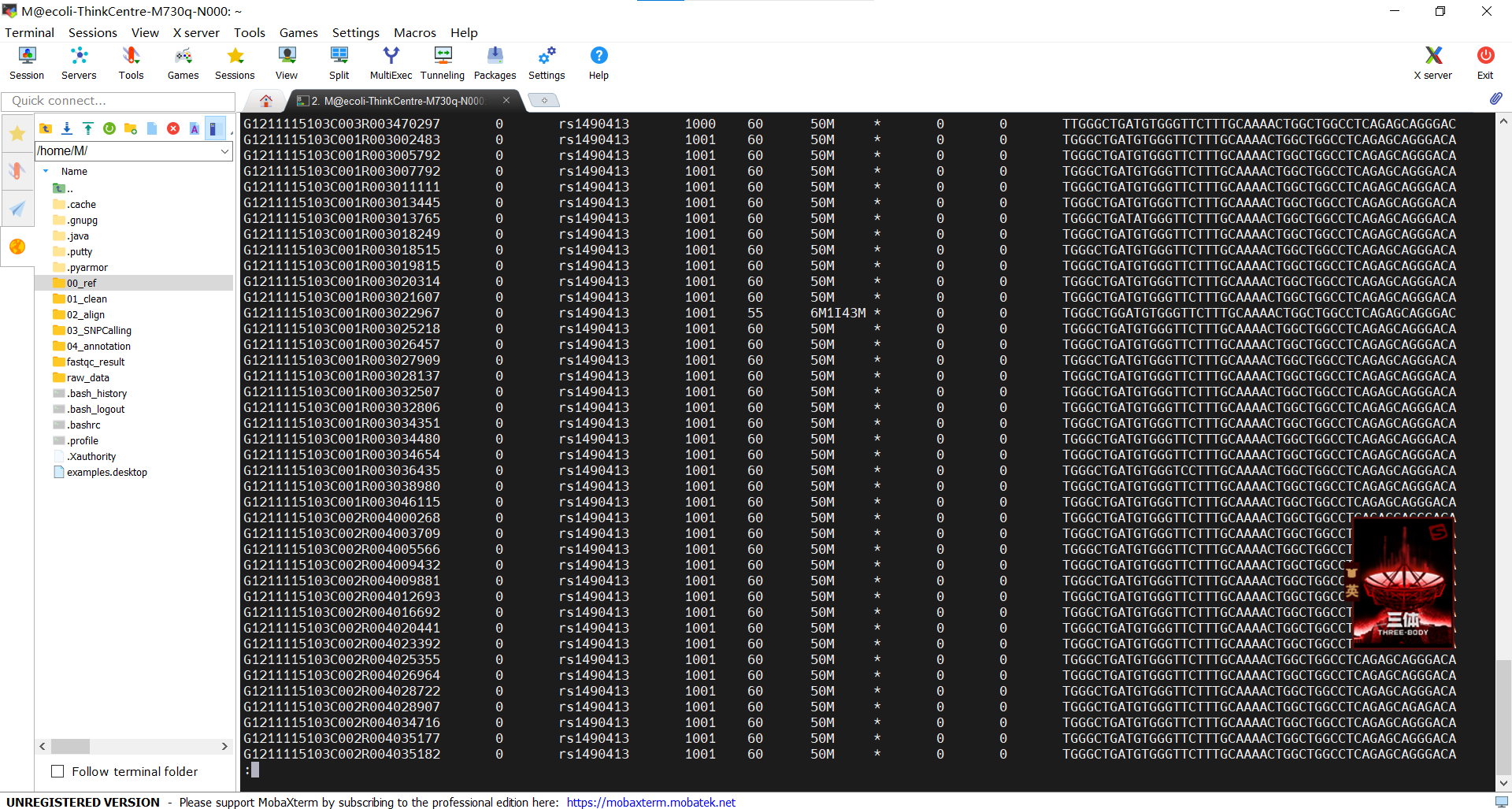
这段BAM文件主要展示了SNP位点rs1490413的部分测序数据结果。1001为染色体位置,60为映射质量,最右面的序列为碱基序列。
# 统计覆盖深度
bamdst -p ./00_ref/target.358.SE50.subSNP.bed -o ./02_align ./02_align/test.clean.sort.uniq.bam
# 查看覆盖深度统计情况
less ./02_align/depth.tsv.gz
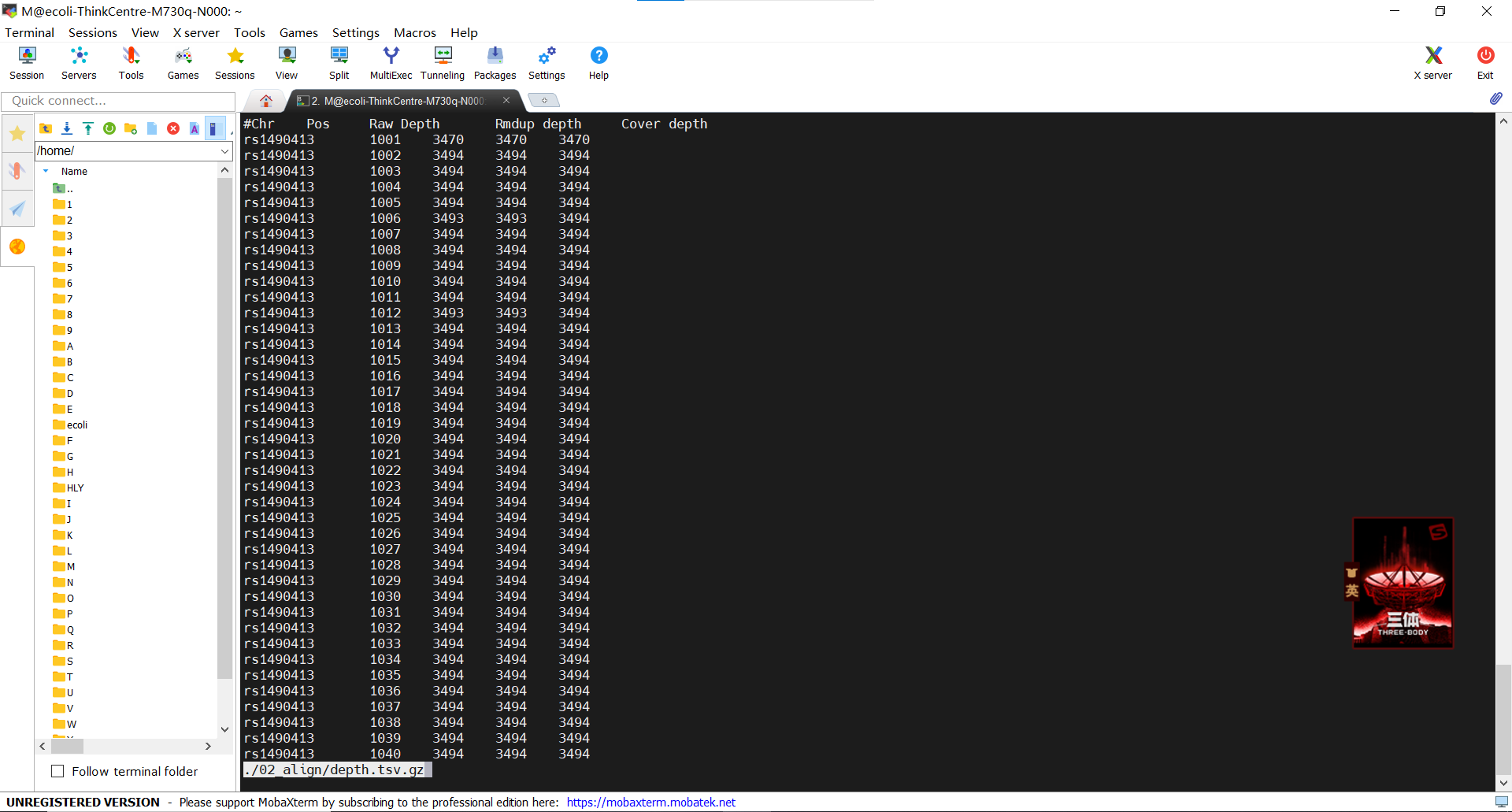
这段文件展示SNP位点rs1490413的覆盖深度统计结果
说明:
Pos: 染色体上位置信息
Raw Depth: 原始深度,即测序数据中特定位置的读取数量
Rmdup Depth: 去除冗余后的深度,排除重复的读取后特定位置的读取数量
Cover Depth: 覆盖深度,表示特定位置被读取覆盖的次数
在最后,小果给小伙伴们总结一下实验的过程:首先使用BWA软件对参考序列建立索引,然后将测序reads与参考序列进行比对。比对结果以SAM格式存储,可以通过Samtools进行查看和处理。为了节约存储空间,将SAM文件转换为BAM文件,并对比对结果进行排序和索引。进一步,统计了覆盖深度,以评估测序数据的质量和覆盖度。 最终,实验结果包括SAM文件,BAM文件,覆盖深度统计数据,以及比对结果的可视化展示和感兴趣的变异位点的查看。
如果小伙伴们平时在生信分析的操作过程中遇到困难,欢迎大家使用小果开发的生信工具平台http://www.biocloudservice.com/home.html,大家在新接触一个知识的时候,与其先花费大量时间死啃知识点,不如先利用好工具先自己上手接触流程,在跑完一遍全流程后再返回去理解知识点,相信可以更好更快地理解,达到事半功倍的效果!比如这一次小果就先把序列比对的操作过程给大家分享出来,帮助大家实际操作一遍后又详细地进行了分步介绍。下一次小果会给大家详细地介绍序列比对的原理,想要进阶提升、成长为生信大师的小伙伴们千万不要错过哦~
另外,为了更好地理解测序片段在参考序列上的位置和变异情况,可以使用IGV软件进行比对结果的可视化,想要学习IGV的小伙伴们记得持续关注小果,小果也会在后续的内容中分享IGV的使用方式哦~