小伙伴们得到GEO数据集原始数据后,发现密密麻麻的基因表达信息不知道从哪里下手。而且基因表达信息往往有很多我们不需要的信息,并且基因表达谱数据往往也是层次不齐。在这里小果自己整理了一份代码,可以把基因表达谱整理出来,并且把探针ID也转换完成,把数据也归一化处理。接下来跟着小果去学习一下吧!
在这里小果使用实例数据GSE63514,平台为GPL-570。小伙伴可以自行在GEO数据库中下载。不过,小果教大家一个小技巧:
在官网搜到自己需要的GEO数据集后,可以在
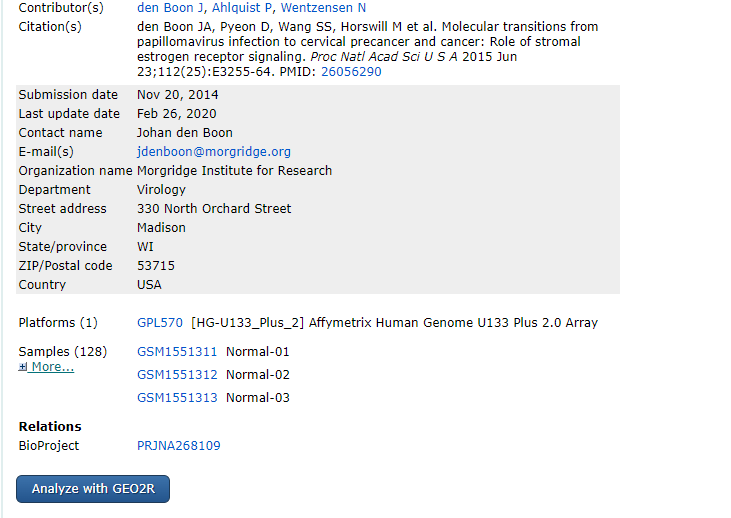
这个选项中选择查看一下这个数据集中含有那些样本:
![]()
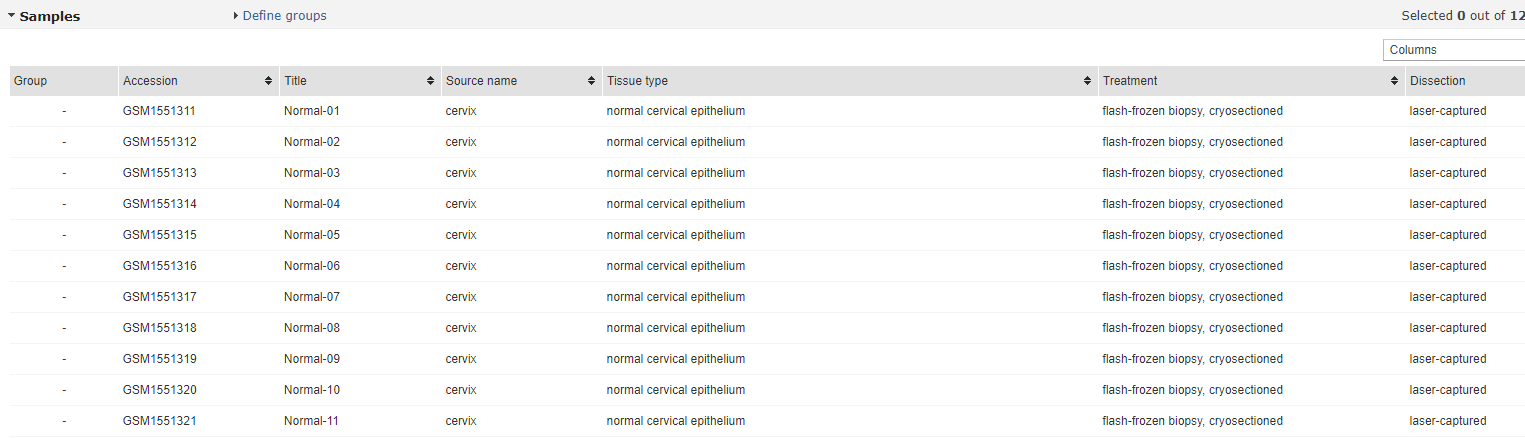
此外,在 
![]()
当我们点击GPL平台后会出现一些信息在这里:

如果这个平台信息含有 ![]() 那么我们处理数据起来就会很方便,利用Gene Symbol就可以转换ID得到我们想要的基因名称。小伙伴们一定要注意,如果没有这个Symbol信息(有的平台可能只会显示Symbol),小果这里的代码可能不太使用。接下来我们就进入正题吧!
那么我们处理数据起来就会很方便,利用Gene Symbol就可以转换ID得到我们想要的基因名称。小伙伴们一定要注意,如果没有这个Symbol信息(有的平台可能只会显示Symbol),小果这里的代码可能不太使用。接下来我们就进入正题吧!
我们得到这个原始数据集,先导入它们:
setwd(“E:\\生信果”)
Sys.setlocale(‘LC_ALL’,’C’)
#读取GPL文件
GPL_table = read.table(‘E:\\生信果\\GPL570-55999.txt’,sep = “\t”,
comment.char = “#”, stringsAsFactors = F,
header = T, fill = TRUE, quote = “”)
#读取GSE文件
GSE4100 <- read.table(‘E:\\生信果\\GSE63514_series_matrix.txt’,sep = “\t”,
comment.char = “!”, stringsAsFactors = F,header = T, fill=TRUE)#43931
#导入文件,我们可以查看一下导入的数据形式。
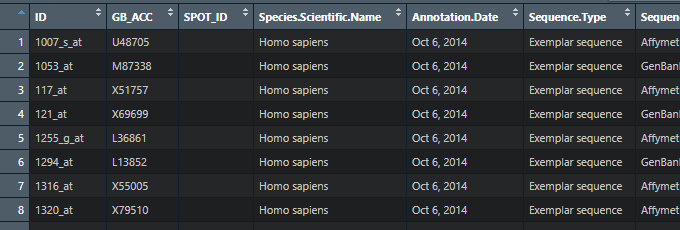
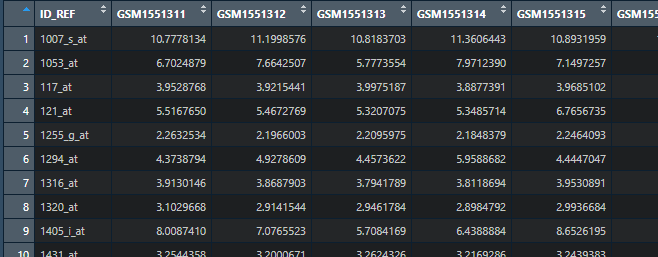
细心的小伙伴会发现,ID这一列不是我们想要的Gene名,这就需要我们吧ID转化一下。
接下来:
ID_Sybmol = GPL_table[,c(1,11)] #GPL对应ID列
colnames(ID_Sybmol)[2]=”Symbol” #更改名称为Symbol,主要是为了对其求平均函数
c(1,11)这里的11是GPL平台表格里面,代表第11列,小果这里GeneSymbol是11列,所以这里是11,小伙伴们需要数一下自己数据Symbol在第几列。
接下来:
#合并ID与基因列
Exp = merge(ID_Sybmol,GSE4100,by.x = “ID”,by.y = “ID_REF”,all=T) #45782个
Exp = Exp[,-1]
View(Exp)#查看一下数据集
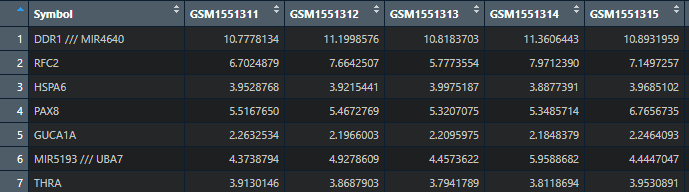
这样一来就得到我们想要的基因名称了,但是又出现一个新的问题,基因名有重复的,而且有个基因含有多个,这该怎么办呢,小果再教一招。对数据过滤:
#数据过滤,对多余的基因进行删除
Exp = Exp[Exp$Symbol != “”,] #
Exp = na.omit(Exp) #
#去除多余的基因,
Exp1 = data.frame(Exp[-grep(“/”,Exp$”Symbol”),]) #去一对多,grep是包含的意思,-就是不包含
write.table(Exp1,”Exp1.txt”,row.names = F,quote = F,sep=”\t”)
那么这里
就得到干净的基因表达谱
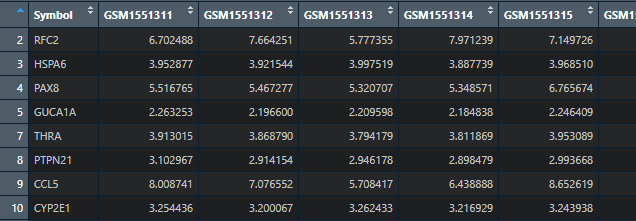
但是,有的小伙伴他的基因表达谱中数据有的异常的大,那么就需要对数据归一化。
接下来小果在继续教大家如何把数据集做平均值处理,这里需要用到for循环处理数据。
#求平均值
meanfun <- function(x) {
x1 <- data.frame(unique(x[,1]))
colnames(x1) <- c(“Symbol”)
for (i in 2:ncol(x)){
x2 <- data.frame(tapply(x[,i],x[,1],mean))
x2[,2] <- rownames(x2)
colnames(x2) <- c(colnames(x)[i], “Symbol”)
x1 <- merge(x1,x2,by.x = “Symbol”,by.y = “Symbol”,all=FALSE)
}
return(x1)
}
Exp2 <- meanfun(Exp1)
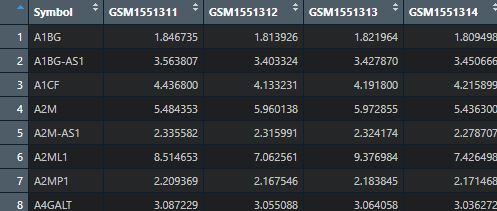 这里数据就会变规整很多。
这里数据就会变规整很多。
我们使用柱状图展示数据形式
# 查看数据
par(cex = 0.7)
n.sample=ncol(Exp2[,-1])
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
boxplot(Exp2[,-1], col = cols,main=”expression value”,las=2)
write.table(Exp2,”Exp_Original.txt”,row.names = F,quote = F,sep=”\t”)
row.names(Exp2) = Exp2[,1]
Exp2 = log(Exp2[,-1])
par(cex = 0.7)
n.sample=ncol(Exp2)
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
boxplot(Exp2, col = cols,main=”expression value”,las=2)
Symbol = row.names(Exp2)
Exp_test = cbind(Symbol,Exp2)
write.table(Exp_test,”Exp.txt”,row.names = F,quote = F,sep=”\t”)
最终得到的数据集就是我们想要的基因表达谱了,小伙伴可以使用得到的基因表达谱做后续的分析啦!
小果这里提醒一下,有的数据集不需要做归一化处理,小伙伴要清楚自己想要得到那个基因表达谱。