大家好,我是小果!今天我要向大家介绍一个神奇的R包——caret,它将帮助您在生物信息学领域发现数据的宝藏。无论您是研究人员还是生物数据分析的爱好者,这个R包都将成为您的得力助手。
Caret(Classification And REgression Training)是一个功能强大的R包,旨在简化机器学习中分类和回归模型的训练和评估过程。它提供了一系列函数和工具,帮助用户轻松地处理数据、选择合适的算法、进行模型训练和预测,并评估模型的性能。
Caret包的主要功能包括:
1. 数据预处理:Caret提供了一系列函数和方法,用于数据的预处理,如数据缩放、数据标准化、缺失值处理等。这些函数可以帮助用户有效地准备数据,以便进行后续的模型训练和评估。
2. 算法选择和训练:Caret支持多种分类和回归算法,包括逻辑回归、支持向量机、随机森林、梯度提升树等。用户可以根据任务的特点和数据的性质选择合适的算法,并使用Caret提供的函数进行模型训练。
3. 模型调优:Caret提供了参数调优的功能,可以帮助用户找到最佳的模型参数组合。它支持自动化的参数搜索和交叉验证,以提高模型的性能和泛化能力。
4. 模型评估:Caret提供了多种性能度量指标,如准确率、精确率、召回率、F1分数等,可以帮助用户评估模型的性能。此外,Caret还提供了可视化工具,如ROC曲线、混淆矩阵等,以帮助用户更好地理解和解释模型的性能。
5. 模型集成和比较:Caret支持模型的集成和比较,用户可以通过集成学习方法(如随机森林、梯度提升树)来提高模型的准确性和稳定性。此外,Caret还提供了模型性能的统计比较方法,用户可以方便地比较不同模型的性能差异。
Caret包的设计旨在提供统一的接口和工作流程,使用户能够更轻松地应用机器学习算法进行分类和回归任务。它减少了繁琐的代码编写和重复的工作,帮助用户更高效地进行数据建模和分析。
总之,Caret是一个强大而灵活的R包,适用于各种机器学习任务。它为用户提供了丰富的功能和工具,使得数据科学家和机器学习从业者能够更加便捷地进行模型的训练、优化和评估。
在大数据时代,我们面对的数据量庞大、复杂多变。如何从中提取有用的信息,揭示生物体的奥秘?使用caret,您将能够探索数据的神秘面纱,揭示隐藏在其中的规律。只需要几行简单的R代码,我就能为您展示caret的神奇之处。小果教一下大家怎么在R语言中使用caret包,利用随机森林,对糖尿病进行预测。具体要如何实现? 今天,小果以代码和提供的数据为例进行演示和说明。
代码具体包括:
Step1 输入数据及划分训练集和测试集
# 这里的”inputdata.txt”是自行准备的本地文件,小果给大家附在最后。
# 以下是一个简单的示例,展示如何使用R包caret进行糖尿病预测模型的训练和评估:
# 安装和加载所需的库
# 安装并导入caret包
install.packages(“caret”)
library(caret)
# 导入数据集以及数据预处理
biological_data <- read.table(“D:/wanglab/life/ziyuan/20230717/inputdata.txt”,header = T)
# 划分训练集和测试集
set.seed(123)
trainIndex <- createDataPartition(1:length(biological_data$diabetes), p = 0.8, list = FALSE)
trainData <- biological_data[trainIndex, ]
testData <- biological_data[-trainIndex, ]
Step2建立模型
# 建立模型
fitControl <- trainControl(method = “repeatedcv”,#method确定多次交叉检验的抽样方法
## 10-fold CV…
number = 10,#number确定了划分的重数
## repeated ten times
repeats = 10)#repeats确定了反复次数
# 模型训练和调优:使用train函数来训练模型,并自动调优模型的参数。
# 通过指定训练数据、目标变量以及之前创建的训练控制参数,Caret将使用交叉验证来训练和评估模型,并自动搜索最佳参数组合。
set.seed(825)
Laplacian <- train(trainData,trainData$diabetes,
method = “rf”,
preProcess = NULL,
weights = NULL,
metric = ifelse(is.factor(y), “Accuracy”, “RMSE”),
trControl = trainControl(),
tuneGrid = NULL,
tuneLength = 20)
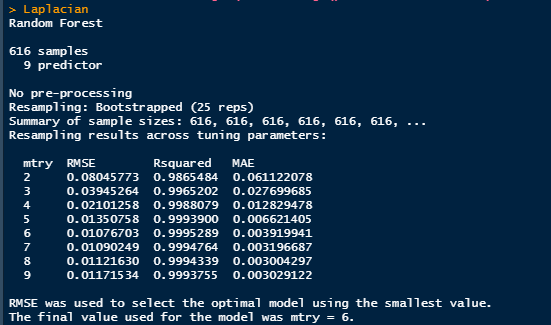
Laplacian
## 采用RMSE法选取最小值的最优模型
## 最终模型使用的参数值为mtry = 6
最优模型
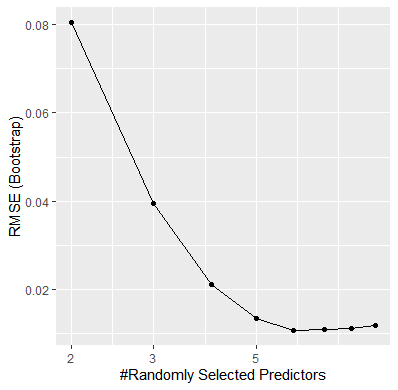
ggplot(Laplacian) + scale_x_log10()
# 数据的曲线图表明,当mtry 大于6时,模型没有变化
最优模型选择的曲线图
Step3进行预测
# 进行预测
predictions <- predict(Laplacian, newdata = testData)# 在测试集上进行预测
print(predictions)# 查看预测结果
# 2.绘制混淆矩阵图
# 提取预测结果
predictions_df <- as.data.frame(predictions)
# 将预测结果和实际值合并为数据框
result_df <- data.frame(Predicted = ifelse(predictions_df$predict > 0.5, “1”, “0”), Actual = as.vector(testData$diabetes))# 转换为二分类预测结果
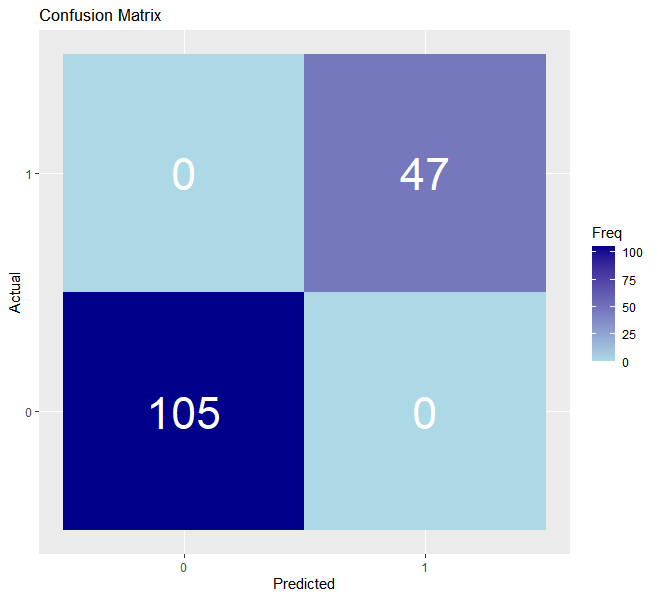
Step4绘制混淆矩阵图
# 绘制混淆矩阵
confusion_matrix <- table(result_df$Predicted, result_df$Actual)
confusion_matrix_plot <- ggplot(as.data.frame(confusion_matrix), aes(x = Var1, y = Var2, fill = Freq)) +
geom_tile() +
geom_text(aes(label = Freq), color = “white”, size = 12) +
labs(title = “Confusion Matrix”) +
xlab(“Predicted”) +
ylab(“Actual”) +
scale_fill_gradient(low = “lightblue”, high = “darkblue”)
# 显示混淆矩阵图
print(confusion_matrix_plot)
混淆矩阵图
Step5绘制AUC曲线图
# 5.绘制AUC曲线图
library(pROC)
roc <- pROC::roc(as.numeric(result_df$Actual), predictions_df$predict)# 计算AUC
plot(roc, main = “Receiver Operating Characteristic (ROC) Curve”,
xlab = “1 – Specificity”, ylab = “Sensitivity”,
print.auc=TRUE,#显示AUC面积
print.thres=TRUE)#添加截点和95%CI)# 绘制AUC曲线
)# 绘制AUC曲线
print(paste(“AUC:”, round(auc(roc), 2)))# 显示AUC值为1
# 请确保将路径”inputdata.txt”替换为实际数据集文件的路径,并根据需要调整模型参数和其他配置。
# 以上代码使用pROC库计算AUC,并使用plot函数绘制AUC曲线。最后,通过调用auc函数获取AUC值,并使用print函数显示在图形上。
# 注意:在绘制AUC曲线之前,确保您的模型输出的是预测的概率值,而不是类别标签。
AUC曲线图

通过上述代码,您将可以轻松构建一个基于随机森林的预测模型,用于解析生物数据中的信息。caret提供了多种机器学习算法和模型评估方法,帮助您选择最合适的模型,并评估其性能。如同探险家一样,您将与caret一同探索数据的未知领域。揭示生物体内潜在的秘密,是一次惊险又有趣的冒险之旅。
但是要更方便得使用生信工具,我不得不向大家推荐我们的云生信平台(http://www.biocloudservice.com/home.html)。。云生信平台是一个全面支持生物信息学应用的平台,为研究人员和医疗专业人员提供高质量的数据分析和解决方案。在云生信平台上,您可以发现更多强大的工具,与其他研究者共享经验,并共同推动生物信息学的发展。
让我们携手前进,利用caret的力量,揭示数据的宝藏,为人类的健康作出更大的贡献!