今天小果为大家带来一款非常新的,但是功能非常强大的生信分析工具– MetaPhlAn 4。
MetaPhlAn是一种计算工具,用于从元基因组霰弹枪测序数据(即非16S)也就是宏基因组数据中分析物种级微生物群落(细菌、古细菌和真核生物)的组成。最新版的MetaPhlAn 4发表在2023年2月23日《Nature Biotechnology》期刊上。MetaPhlAn 4利用已知微生物基因组和MAGs的组合扩展集,定义了物种水平上的基因组箱(SGBs),以进行更全面的宏基因组分类学分析。从101万个原核生物参考基因组和宏基因组组装基因组的集合中,在物种水平为26970个基因组箱定义了特异性的标记基因,其中4992个在物种水平上尚未分类。MetaPhlAn 4在全球人类肠道微生物中增加了约20%的短序列物种注释,而在尚未充分研究的环境(如反刍动物瘤胃微生物组等)中增加了40%。MetaPhlAn 4在综合评估分析方面比现有可替代方案更准确,同时还可以对未培养分离的生物体进行准确定量。总而言之,MetaPhlAn 4是宏基因组物种注释中一个方便而且准确的最新工具。
接下来小果就为大家带来MetaPhlAn 4的安装和使用教程,下面的操作都要在服务器上进行,如果没有服务器,那小果也没法啦。
一、创建conda环境
conda create –name mpa -c bioconda python=3.7
创建一个名叫mpa的环境
二、安装metaphlan4
source activate mpa
conda install -c bioconda metaphlan
这一步可能会安装很久,有时候可能会出现安装不上的问题,这时候我们可以用pip来安装。
三、安装数据库
metaphlan –install
这是最麻烦的一步(一般用这个命令都安装不上),下载速度较慢且总共20多G,建议去官网下载,下载最新的那个数据库。
这六个文件建议都下载,网址分别是:
http://cmprod1.cibio.unitn.it/biobakery4/metaphlan_databases/
http://cmprod1.cibio.unitn.it/biobakery4/metaphlan_databases/bowtie2_indexes/
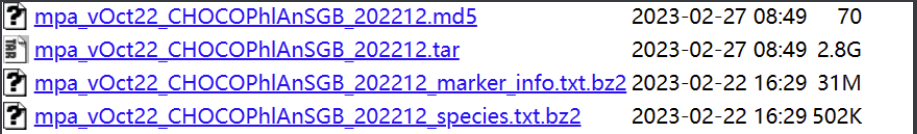

这几个文件全部都要放在metaphlan_databases这个文件夹里面,路径一般是这个:
path/to/anaconda3/envs/mpa/lib/python3.7/site-packages/metaphlan/metaphlan_databases
注意我前面这个path/to/是大家放anaconda3的文件路径。
后面运行metaphlan4跑一个数据就会自动建库。建库后的metaphlan_databases就有这些文件:
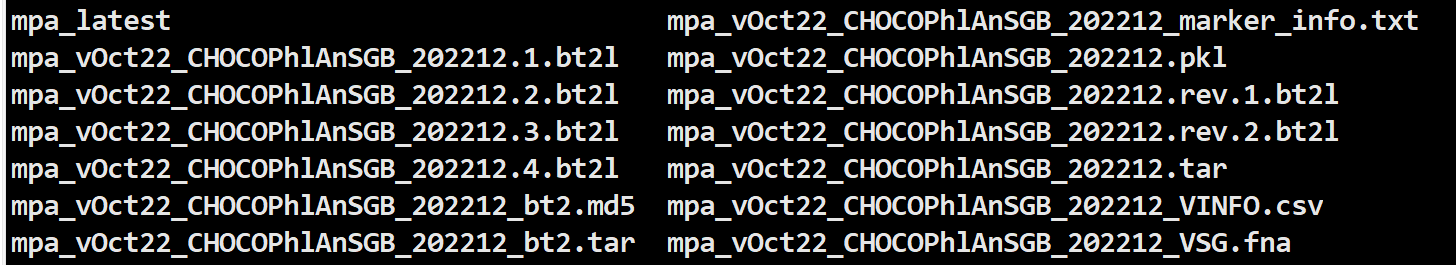
注意mpa_latest这个文件,如果您没有的话,可以自己vim写一个,里面的内容也就是下载的数据库的名字:

四、运行
双端数据:
metaphlan *.fastq1,*.fastq2 –input_type fastq -o *.txt –nproc 10 –stat_q 0.1 –bowtie2out bowtie2out_*.bz2
也可以单端,就只输入一个fastq数据,注意fastq1和fastq2之间有个逗号。
其中参数的含义:
–input_type输入数据类型
–stat_q越低假阳性率越高,默认是0.2,但是可以按照数据具体情况调整,0.1应该是可以接受的
–nproc线程
五、数据合并截取
多个数据运行完成后,需要对多个结果进行合并(不同版本的数据不能合并) :
merge_metaphlan_tables.py metaphlan_output*.txt > output/merged_abundance_table.txt
#筛选出不同水平物种丰度表格
grep -E ‘(s__)|(clade_name)’ merged_abundance_table.txt |grep -v ‘t__’|sed ‘s/^.*s__//g’|sed ‘s/\ \ /\ /g’|sed ‘s/\ /\t/g’ > merged_abundance_table_species.txt
grep -E ‘(g__)|(clade_name)’ merged_abundance_table.txt |grep -v ‘s__’|sed ‘s/^.*g__//g’|sed ‘s/\ \ /\ /g’|sed ‘s/\ /\t/g’ > merged_abundance_table_genus.txt
grep -E ‘(f__)|(clade_name)’ merged_abundance_table.txt |grep -v ‘g__’|sed ‘s/^.*f__//g’|sed ‘s/\ \ /\ /g’|sed ‘s/\ /\t/g’ > merged_abundance_table_family.txt
grep -E ‘(o__)|(clade_name)’ merged_abundance_table.txt |grep -v ‘f__’|sed ‘s/^.*o__//g’|sed ‘s/\ \ /\ /g’|sed ‘s/\ /\t/g’ > merged_abundance_table_order.txt
grep -E ‘(c__)|(clade_name)’ merged_abundance_table.txt |grep -v ‘o__’|sed ‘s/^.*c__//g’|sed ‘s/\ \ /\ /g’|sed ‘s/\ /\t/g’ > merged_abundance_table_class.txt
grep -E ‘(p__)|(clade_name)’ merged_abundance_table.txt |grep -v ‘c__’|sed ‘s/^.*p__//g’|sed ‘s/\ \ /\ /g’|sed ‘s/\ /\t/g’ > merged_abundance_table_phylum.txt
六、脚本
大家可以一个一个数据的跑,当然也可以写个循环,这样效率更高,这个是小果自己写的一个最简单的shell脚本:
for i in `ls data/split`; do
metaphlan data/${i}_1.fq.gz,data/${i}_2.fq.gz –input_type fastq –nproc 30 –output metaphlan/${i}.txt –bowtie2out metaphlan-1/${i}.bowtie –stat_q 0.05; done
###split文件夹里面是所有数据名称的文件
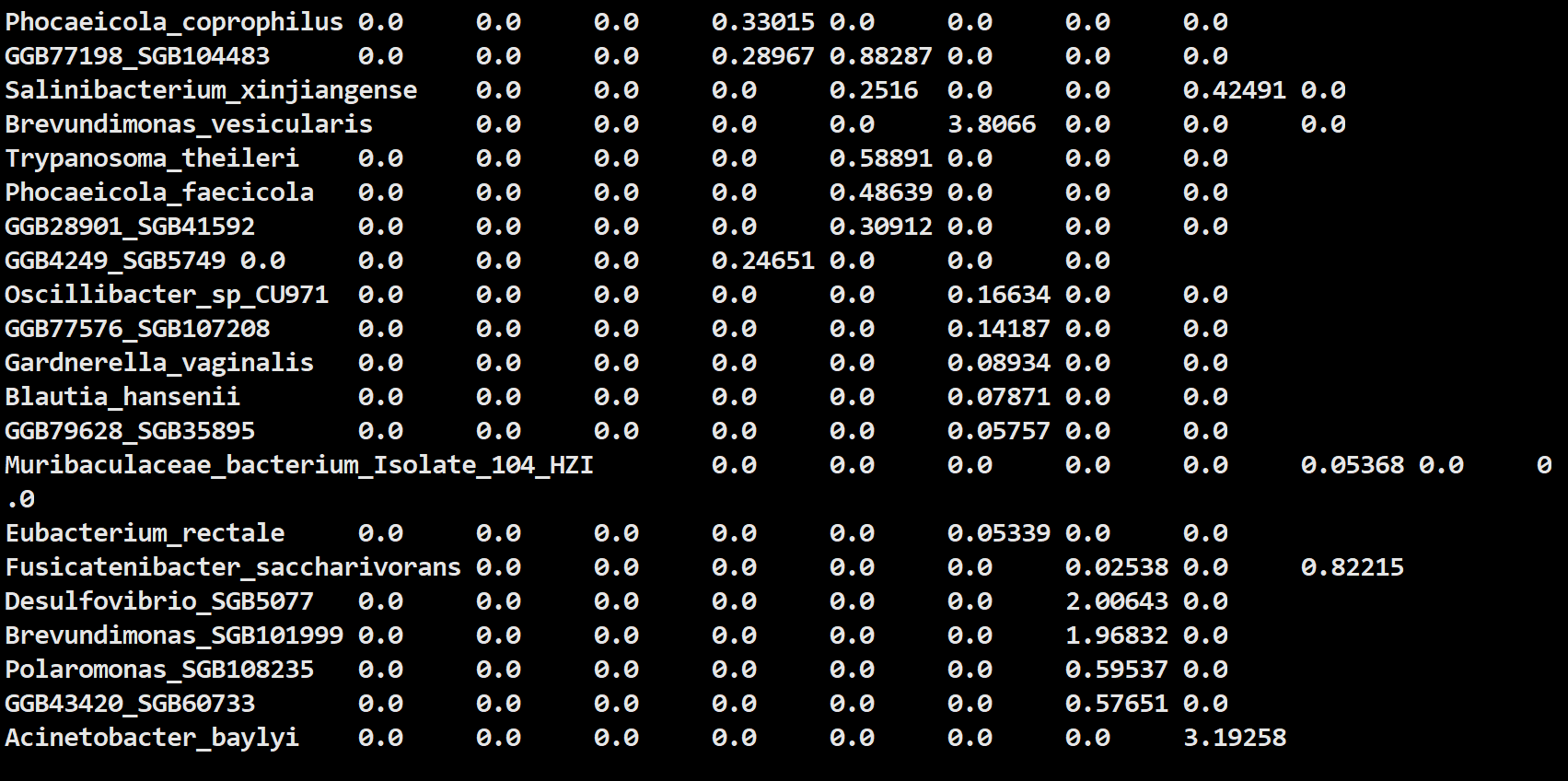
运行完的结果大概是这样:
这就是本次教程的全部内容啦,后续小果会为大家带来更多的宏基因组和扩增子软件的使用教程。