不知道有多少刚开始接触单细胞测序分析的小伙伴在面对无穷无尽的R包和代码时感到畏难,仿佛一堆知识要强行涌入我们的脑海。今天小果想向大家介绍一个R包——popsicleR。该R包的作者整合了单细胞转录组分析所需的R包,用七行代码就可以完成从质控到细胞注释的整套流程。学习使用该R包可以为初学者构建思维框架,从而使学习过程更加简单。
下面,小果将为大家详细介绍一下popsicle。它结合了广泛使用的流程中实现的方法,以交互方式执行 scRNA-seq 数据分析的所有主要预处理和 QC 步骤。该软件包由七个功能组件组成:
Step 1:执行质量控制指标的探索
Step 2:细胞筛选
Step 3:双细胞检测
Step 4:数据标准化
Step 5:缩放及回归
Step 6:细胞聚类
Step 7:细胞注释
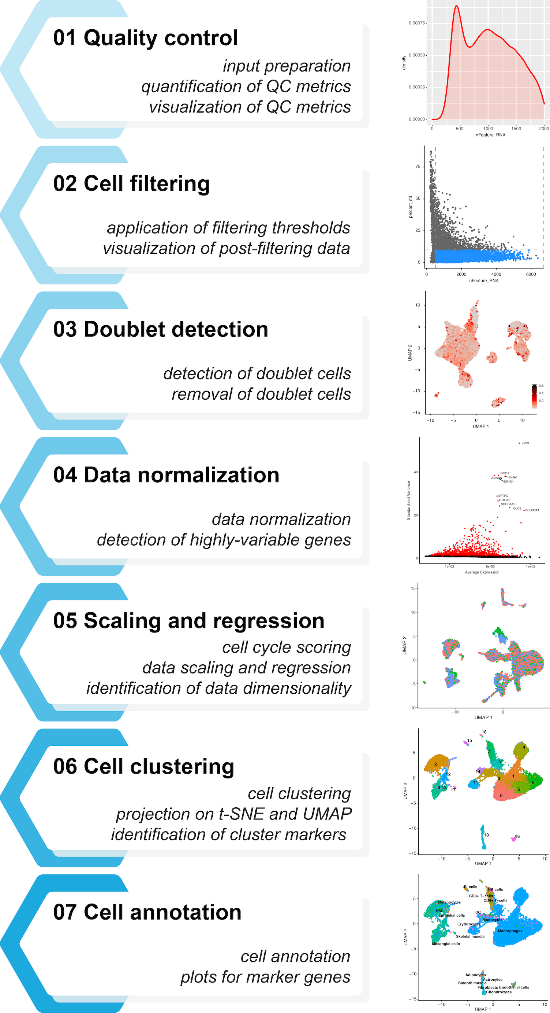
在分析的每个步骤中,popsicleR 通过彩色文本消息交互式地指导用户,并在专用文件夹中保存各种图,以研究多个 QC 指标并评估过滤和回归参数对细胞群识别和分类的影响。跑完流程后可为小伙伴们产出20多张各式各样且美观的图表。
小果这边已经为小伙伴们准备好输入文件了,获取链接放在文章的最后哦~就让我们正式开始今天的学习吧!
# Step 0 安装popsicleR包及其依赖
因为popsicleR整合大量的R包,因此在安装依赖的过程中可能有一些繁琐哦,下面的代码虽然很多,但只需要小伙伴们直接运行就可以啦。
options(timeout = 999) # 方便在GitHub上下载对应的R包
CRANdep <- c(“Seurat”,”reticulate”,”R.utils”,”dplyr”,”ggplot2″,”clustree”,”ape”,”gtools”,
“future”,”grid”,”gridExtra”,”magrittr”,”limma”,”patchwork”,
“crayon”,”ggExtra”,”RColorBrewer”,”ggplotify”,”RANN”,”umap”,
“celldex”,”curl”,”httr”,”lattice”,”shinythemes”,”usethis”,”rcmdcheck”,
“roxygen2″,”rversions”,”devtools”,”pheatmap”,”BiocManager”,”corrplot”)
newPackages <- CRANdep[!(CRANdep %in% installed.packages()[,”Package”])]
if(length(newPackages)){install.packages(newPackages)}
CRANarcdep <- c(“Matrix”,”optimbase”,”optimsimplex”,”neldermead”,”session”)
newPackages <- CRANarcdep[!(CRANarcdep %in% installed.packages()[,”Package”])]
if(length(newPackages)){
packagesurl <- c(“https://cran.r-project.org/src/contrib/Archive/Matrix/Matrix_1.3-2.tar.gz”,
“https://cran.r-project.org/src/contrib/Archive/optimbase/optimbase_1.0-9.tar.gz”,
“https://cran.r-project.org/src/contrib/Archive/optimsimplex/optimsimplex_1.0-7.tar.gz”,
“https://cran.r-project.org/src/contrib/Archive/neldermead/neldermead_1.0-11.tar.gz”,
“https://cran.r-project.org/src/contrib/Archive/session/session_1.0.3.tar.gz”)
for (i in 1:length(newPackages)){
source_repo <- packagesurl[grep(newPackages[i], packagesurl)]
install.packages(source_repo, repos=NULL, type=”source”)
}
}
BioCdep <- c(“SingleR”,”limma”,”BiocFileCache”,”AnnotationHub”,
“ExperimentHub”,”celldex”,”scDblFinder”)
newPackages <- BioCdep[!(BioCdep %in% installed.packages()[,”Package”])]
if(length(newPackages)){BiocManager::install(newPackages)}
if(!”scMCA”%in% installed.packages()[,”Package”]){devtools::install_github(“ggjlab/scMCA”)}
devtools::install_github(“bicciatolab/popsicleR”)
library(popsicleR)
载入R包后出现
即为安装成功,我们便可以进行下一步啦
# step 1 从原始数据创建 Seurat 对象并可视化 QC 指标
seurat_obj <- PrePlots(sample = ‘CRR073027’,
input_data = ‘filtered_feature_bc_matrix/’,
percentage = 0.1, gene_filter = 200, cellranger = TRUE, organism = “human”)
@para sample: 研究项目的name;
@para input_data: 输入文件的路径,在这面小伙伴们既可以使用小果为大家准备好的cellranger的矩阵结果,也可以是非cellranger的普通计数矩阵,都是兼容的哦
@para genelist: 在这里如果小伙伴们有属于自己的一套与细胞对应的marker列表也可以使用向量的形式进行添加哦,小白没有的话,也没有关系,作者为大家准备好了一套marker。
@para percentage: 这里限制了所有的基因起码在1%的细胞中进行表达,默认值为0.1
@para gene_filter: 限制所有的细胞最起码检测出200个基因
@cellranger: 若输入文件为cellranger的输出文件则设定为TRUE
@organism: 选择研究的物种
下面小果为大家筛选了运行完质控结果后的数张结果图给各位小伙伴预览一下
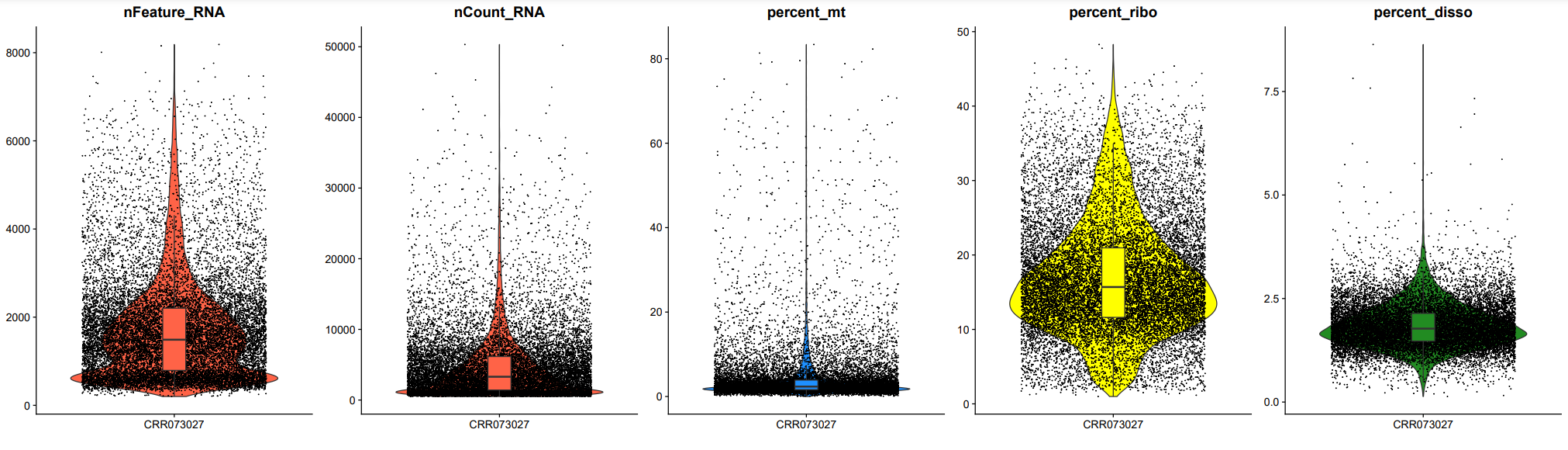

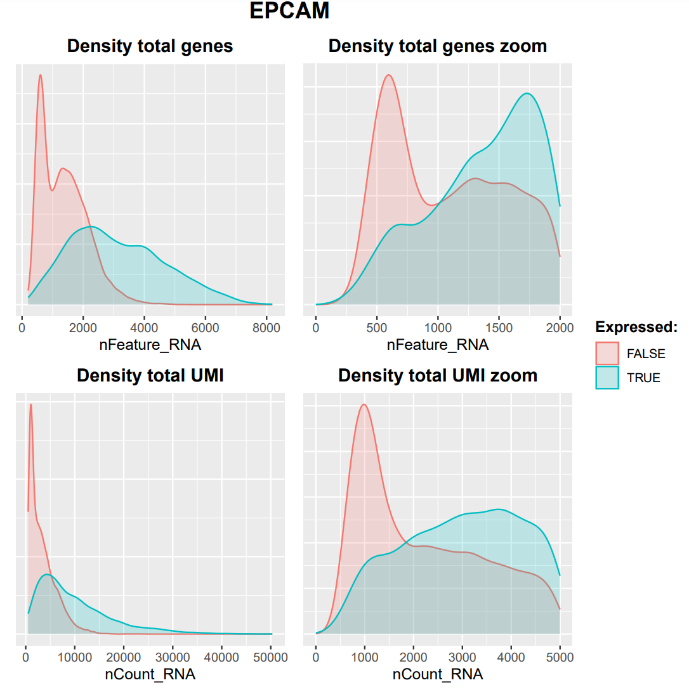
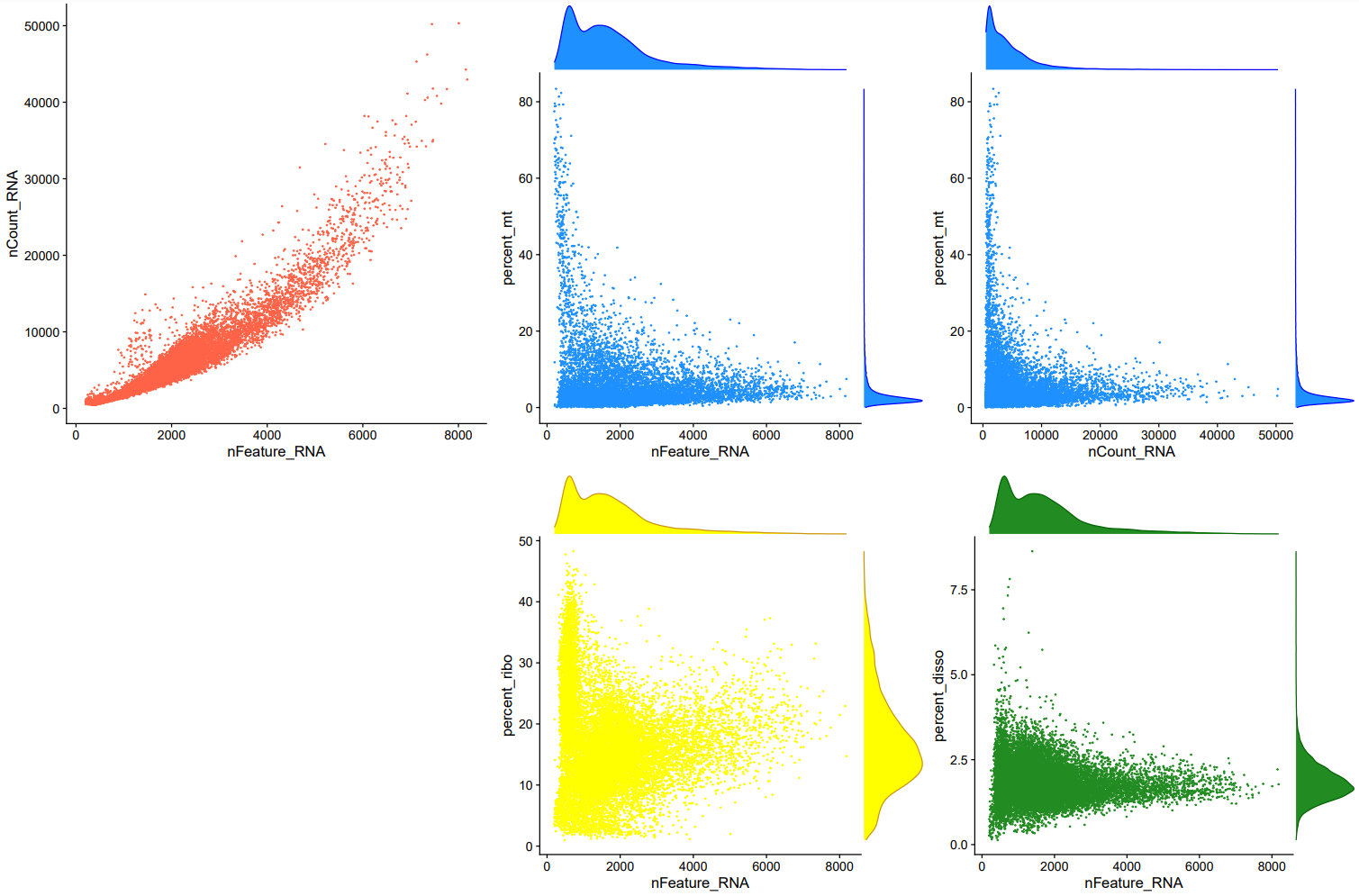
# step 2 双重细胞检测
该处代码的运行时间会较长,有条件的小伙伴建议使用服务器进行运行哦~
seurat_obj <- CalculateDoublets(UMI = seurat_obj, method = “scrublet”, dbs_thr =’none’, dbs_remove = FALSE)
# step 3 双重细胞移除
seurat_obj <- CalculateDoublets(UMI = seurat_obj, method = “scrublet”, dbs_thr = 0.22, dbs_remove = TRUE)
# 参数说明:
@para UMI: 输入的单细胞转录组数据对象
@para method: 双重细胞检测方法,可以选择”scrublet”,”scDblFinder”其中一种方式
# 来自小果的温馨提示,dbs_thr参数是在选择scrublet方法的情况下才进行选择的哦
@para dbs_thr: 设定的双重细胞阈值,这里设置为’none’
@para dbs_thr: 设定的双重细胞阈值,这里设置为0.22
@para dbs_remove: 是否移除双重细胞
# step 4标准化
seurat_obj <- Normalize(UMI = seurat_obj, variable_genes = 2000)
# 参数说明:
# @para UMI: 输入的单细胞转录组数据对象
# @para variable_genes: 选择的变异基因数量,这里设置为2000
# step 5 回归
seurat_obj <- ApplyRegression(UMI = seurat_obj, organism = “human”, variables = “none”, explore_PC = FALSE)
# step 6对PC值进行观测后进行回归研究
seurat_obj <- ApplyRegression(UMI = seurat_obj, organism = “human”, variables = “none”, explore_PC = TRUE)
# 参数说明:
@para UMI: 输入的单细胞转录组数据对象
@para organism: 研究的物种,这里选择了”human”
@para variables: 进行回归分析的变量,这里设置为”none”
# 在我们探索主成分的个数后便可对该参数进行调整
@para explore_PC: 是否探索主成分,这里设置为FALSE
@para explore_PC: 是否探索主成分,这里设置为TRUE
# step 7细胞类型注释
seurat_obj <- MakeAnnotation(UMI = seurat_obj, organism = “human”, marker.list = “none”, cluster_res= 0.8)
# 参数说明:
@para UMI: 输入的单细胞转录组数据对象
@para organism: 研究的物种,这里选择了”human”
@para marker.list: 细胞类型标记基因列表,这里设置为”none”
@para cluster_res: 聚类分辨率,这里设置为0.8
可以看出,虽然这段代码有7行,但实际上有两行代码是为了重复运行以选择参数的,因此我们可以只用5行代码就完成单细胞转录组分析。
popsicleR这个R包的源代码将近有2000行,感兴趣的小伙伴可以进入官方文档进行学习哦(https://github.com/bicciatolab/popsicleR/),这不仅仅是对单细胞转录组分析流程的学习,还可以通过阅读源码了解到各种各样的表格的绘制代码,在日后的研究学习中颇有益处。
好啦~今天小果的分享就到这里啦!希望今天小果的分享可以帮到刚开始入门单细胞转录组分析的小伙伴们哦~如果小伙伴有其他数据分析需求,可以尝试使用本公司新开发的生信分析小工具云平台,零代码完成分析,非常方便奥,云平台网址为:(http://www.biocloudservice.com/home.html),其中也包括了通路表达分析(http://www.biocloudservice.com/313/313.php),单细胞的基因共表达分析(http://www.biocloudservice.com/906/906.php)等各种小工具哦~,有兴趣的小伙伴可以登录网站进行了解。