10×genomics因其分选细胞效果好,通量高等优点已成为主流的单细胞测序平台。
小果上期对它的原理进行了介绍,感兴趣的小伙伴可以去看。这期小果将介绍10×genomics单细胞测序分析环境的搭建与用10×平台官方出的数据处理流程Cell Ranger进行数据处理与count表达矩阵的生成。
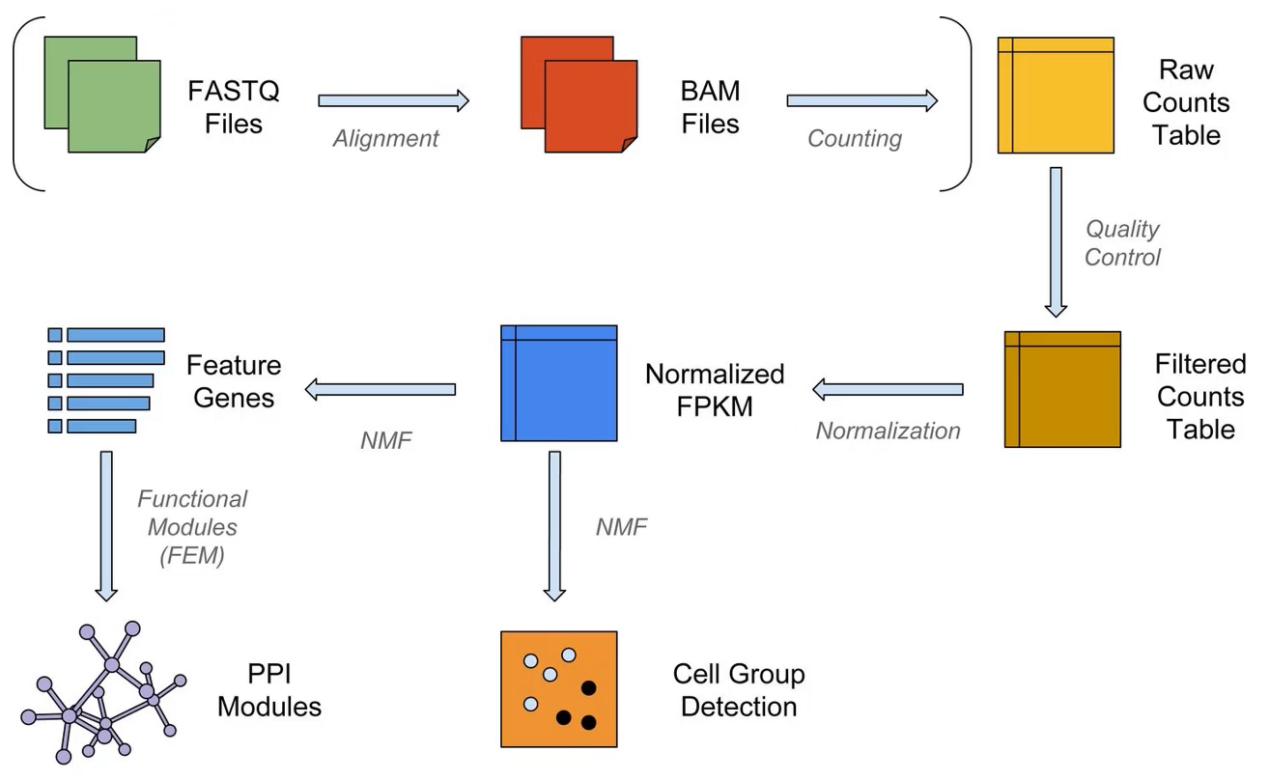
通常这是单细胞分析的整个流程。
与许多组学分析一样,先将FASTQ文件比对,转换为二进制的BAM文件,然后进行基因表达计数。拿到表达矩阵后才能进行下一步的分析。
今天小果带大家完成流程的搭建与上游的的数据处理。就是这张图上面三个步骤。
一、准备工作
(一)软件安装
1.CellRanger
Cell Ranger是10×genomics官方出的数据处理分析工具,它的适配度好,功能强大,操作简单等优点,成为了单细胞测序数据分析的必备工具。
在获得单细胞的测序数据后,使用Cell Ranger来进行数据的处理与分析的工作流程。
What is Cell Ranger? -Software -Single Cell Gene Expression -Official 10x Genomics Support
上面是10×genomics测序平台Cell Ranger主页链接,详细介绍了Cell Ranger是什么与怎么用。
下载需要填写信息完成注册。
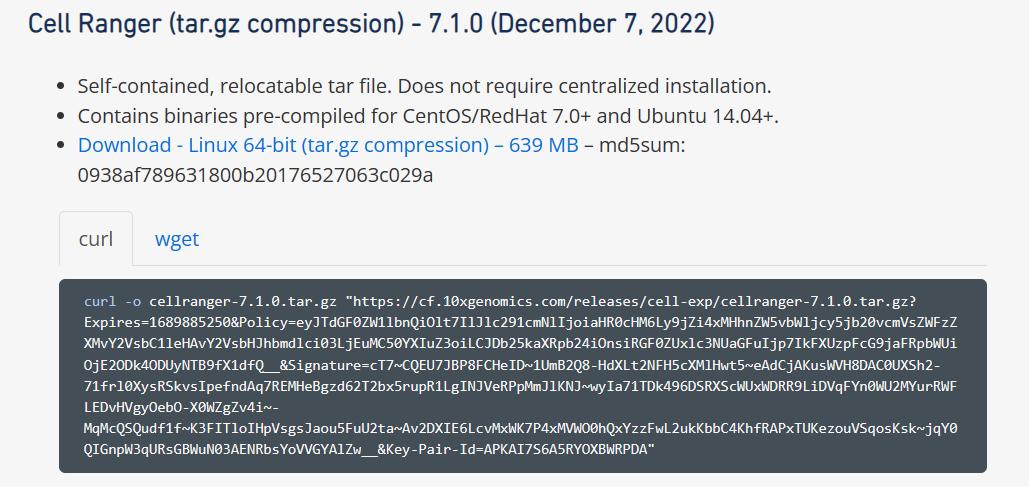
复制下载链接到linux命令行,运行并下载
#随后解压缩下载的软件包
tar -zxvf cellranger-7.1.0.tar.gz
在命令行敲一下,ok,软件安装好了。
Loupe Browser
Loupe Browser是一个桌面应用程序,提供交互式可视化分析来自不同 10x 基因组学解决方案的数据的功能。它能轻松查询 10x 基因组学数据的不同视图快速深入了解数据。 
下载仍然需要填写一次信息。填写好信息后便可以下载。
在windows系统下打开安装包,进行安装即可。
这是应用的界面
3.bcl2fastq
是illumina平台内置的一个软件,Cell Ranger没有集成这个软件,我们需要自己去下载。有了这个软件之后,Cell Ranger可以完成从bcl测序文件,到最后结果生成,一条龙式地完成。
bcl2fastq and bcl2fastq2 Conversion Software Downloads (illumina.com.cn)
bcl2fastq的网站
同样需要注册填写信息,就可以进一步安装了
https://support.illumina.com.cn/content/dam/illumina-support/documents/downloads/software/bcl2fastq/bcl2fastq2-v2-18-0-12-tar.zip
这里贴上下载的地址
#解压缩
unzip bcl2fastq/bcl2fastq2-v2-18-0-12-tar.zip
tar -zxvf bcl2fastq/bcl2fastq2-v2-18-0-12-tar.gz
#编译软件
cd bcl2fastq2
chmod ugo+x src/configure
chmod ugo+x src/cmake/bootstrap/installCmake.sh
mkdir bin
cd bin
../src/configure –prefix=${PWD}/bin
make
make install
4.创建conda环境安装软件
#安装mamba
conda install -y mamba
#创建虚拟环境
conda crate -n -y singlecell
#激活环境
conda activate singlecell
#下载软件
mamba install -y fastqc
mamba install -u multiqc
mamba install -y fastp
mamba install -y star
mamba install -y bioconductor-singler
mamba install -y samtools
mamba install -y featurecounts
mamba install -y r-seurat
mamba install -y umi_tools
数据下载
参考序列下载
#人基因组
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz
#小鼠基因组
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz
#人和小鼠混合
wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-and-mm10-2020-A.tar.gz
#Sample Index Set Sequences
wget https://cf.10xgenomics.com/supp/cell-exp/chromium-shared-sample-indexes-plate.csv
wget https://cf.10xgenomics.com/supp/cell-exp/chromium-single-cell-sample-indexes-plate-v1.csv
wget https://cf.10xgenomics.com/supp/cell-exp/gemcode-single-cell-sample-indexes-plate.csv
2.练习数据下载
#pbmc 1k数据
wget https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_v3/pbmc_1k_v3_fastqs.tar
1000个单细胞的测序数据
使用cellranger分析单细胞数据
Basecalling
1.将测序原始数据bcl文件转换为fastq文件
#案例数据,illuminca测序原始数据
wget https://cf.10xgenomics.com/supp/cell-exp/cellranger-tiny-bcl-1.2.0.tar.gz
wget https://cf.10xgenomics.com/supp/cell-exp/cellranger-tiny-bcl-simple-1.2.0.csv
tar -zxvf cellranger-tiny-bcl-1.2.0.tar.gz

这是这个原始测序数据所包含的内容,数据在这个Data目录下面。

simple文件是个表格。这个案例数据只有一个样本。1代表第一个样本,然后样本名,最后是Index号码。
cellranger mkfastq –id=output –run=cellranger-tiny-bcl-1.2.0 –csv=cellranger-tiny-bcl-simple-1.2.0.csv –localcores=12 –localmem=32
使用cellranger mkfastq软件
–id: 输入的文件夹
–run: 输入的文件
–csv: 数据的列表
–localcores: 控制CPU核心数
–localmem: 控制内存数目
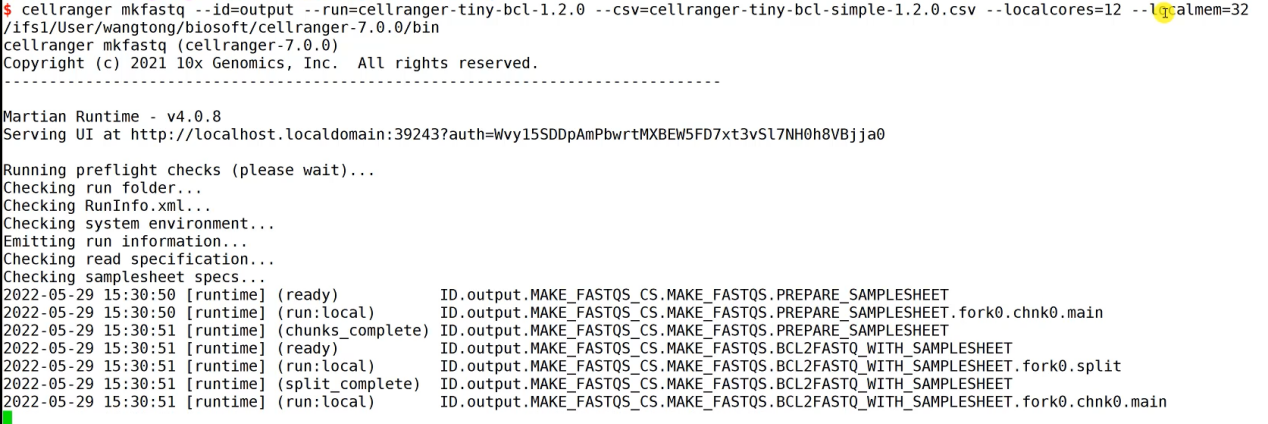
非常简单,只要输入文件,及输出的文件夹就行了。因为数据很小,很快就运行好了。
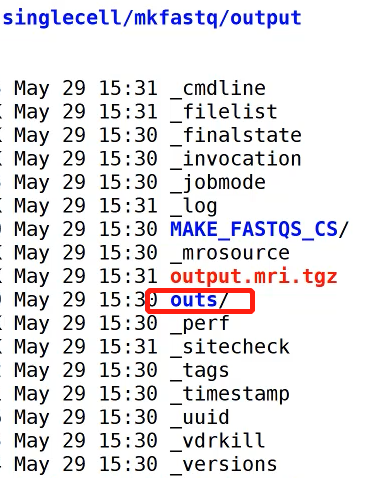
生成的fastq就在这个文件夹里。
Test_sample就是最终的结果。文件名会根据simple文件的名称命名。
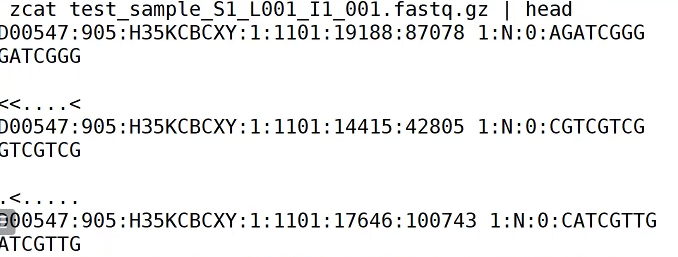
打开第一个看一下,其为Index序列。
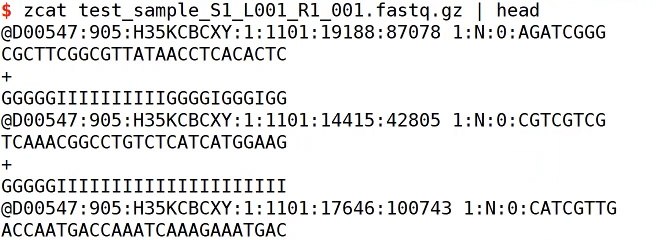
Read1为barcod+UMI
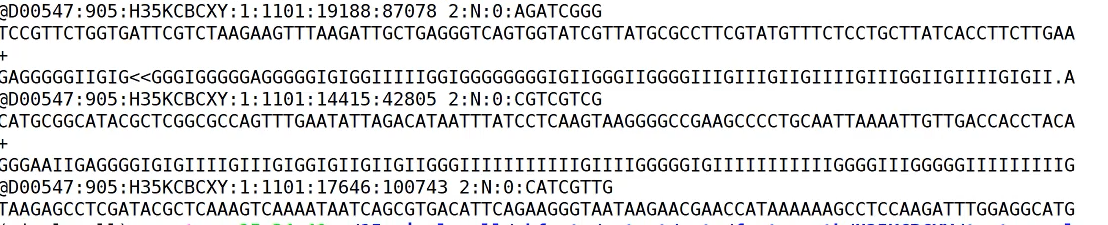
Read2是真正的我们想要的测序序列。
该部分为演示如何将bcl转化为fastq。
质控
接下来我们对练习数据进行处理,这里我们打开案例的数据。使用通配符*匹配所有fastq的read2。然后进行质控。
#对read2 进行质控
ll pbmc_1k_v3_fastqs/pbmc_1k_v3*_R2_001.fastq.gz
fastqc -t 12 -f fastq -o qc1 pbmc_1k_v3_fastqs/pbmc_1k_v3*_R2_001.fastq.gz
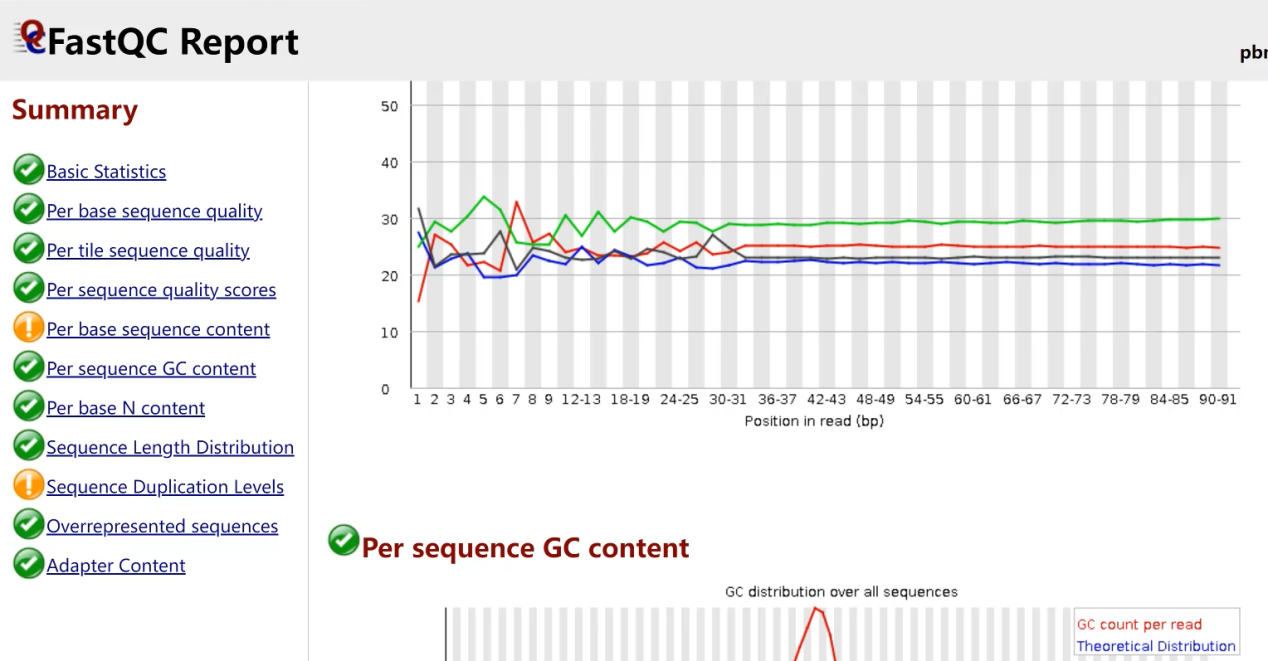
质控结果,显示测序数据质量较好。
10×genomics的测序数据通常质量较好,所以绝大部分不用进行质控以及过滤。
实在过滤的话呢,由于read1和read2不一样,前者是barcode,后者是测序数据,对测序数据过滤掉以后,二者索引不匹配,引起操作麻烦。
生成表达矩阵
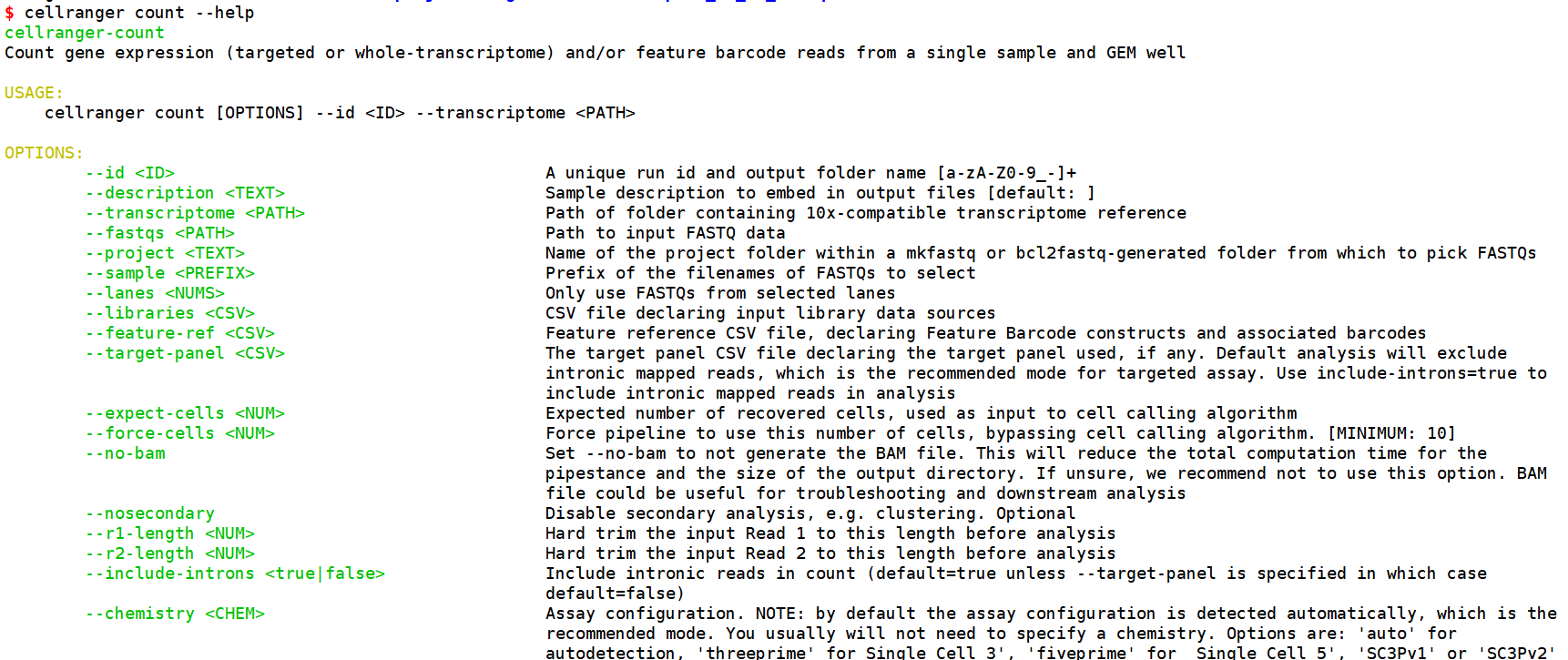
Cellranger count的选项参数,该软件主打的就是一个简单方便,用起来简单绝对是个优点。
输入前面三个参数就可以工作了。
#计算count 矩阵,pbmc 1000个细胞,等号之间不要有空格
cellranger count –id=run_count_1kpbmcs –fastqs=pbmc_1k_v3_fastqs –sample=pbmc_1k_v3 \
–transcriptome=refdata-gex-GRCh38-2020-A/ \
–localcores=12 –localmem=32
设置输入输出,剩下的按照默认的就行了。
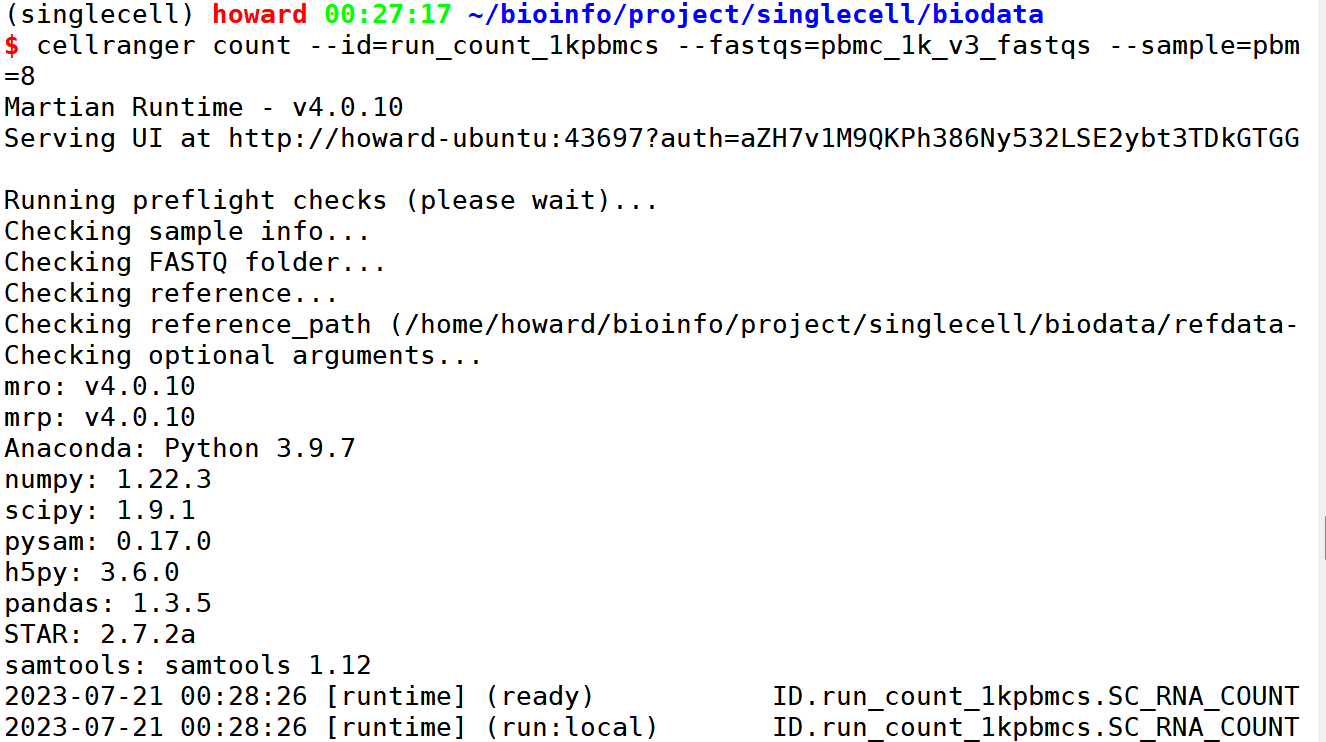
这样它就运行起来了。
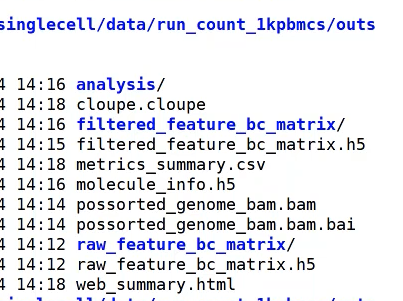
最终的结果在outs里面
小果今天的讲解就到这里了~下期我将会带来对单细胞测序分析结果的解读~我们不见不散
单细胞测序分析内容欢迎尝试本公司开发的云平台生物信息分析工具,零代码完成分析,云平台网址:http://www.biocloudservice.com/home.html