上一期小果用loupe brosew进一步探索单细胞测序处理结果,但是它的功能有限,还无法实现复杂的功能。如果要做更深入的分析是还不够的。所以今天隆重向大家介绍,Seurat——一个专业做单细胞测序下游分析的R包。

在上游地处理中Cell Ranger生成了feature barcode矩阵,并进行了基本的背景过滤,但是还远远不够。单细胞测序更深入的挖掘可以用R包Seurat。
Tools for Single Cell Genomics • Seurat (satijalab.org)
它功能强大可以进一步地实现:
- 进一步过滤
- 聚类,分细胞亚群
- 标记基因
- 可视化,每一步都可以进行可视化
- 无缝读取10×数据
- ….

该包在单细胞测序下游分析领域非常著名与权威,其发布地每个版本都有高水平文章发布。
下面我们通过一个案例来开始学习吧~
安装与加载
install packages(“Seurat”)
library(dplyr)
library(patchwork)
加载案例数据
在本教程中,我们将分析10X Genomics免费提供的外周血单核细胞(PBMC)数据集。有2700个单细胞在Illumina NextSeq 500上进行了测序。
https://cf.10xgenomics.com/samples/cell/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz
原始数据下载链接。
pbmc.data <- Read10X(data.dir = “pbmc3k/filtered_gene_bc_matrices/hg19/”)
使用Read10X函数读取10×genomics 平台cellranger pipline的输出,返回唯一的计数矩阵。该矩阵中的值表示在每个细胞(列)中检测到的每个特征(即基因,行)的分子数量。
生成Seurat对象
pbmc <- CreateSeuratObject(counts = pbmc.data, project = “pbmc3k”, min.cells = 3, min.features = 200)
接下来,我们使用CreateSeuratObject来创建Seurat对象。该对象充当一个容器,其中包含单细胞数据集的数据(如计数矩阵)和分析(如PCA或聚类结果)。
min.cells = 3,是指每个细胞中UMIs的个数最小是3
min.features = 200,是指每个细胞中基因数目最少为200,否则会被过滤掉。
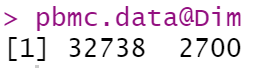
查看数据集维度,有32738个基因,2700个细胞。
数据质控与过滤
对于现在的数据我们仍然需要进一步的过滤。来去除空细胞,与死细胞。
那么我们该如何判断一个细胞是死细胞还是空细胞呢?我们有对应之策,如果是空细胞的画那么捕获到的umi数量就会很少。如果是死细胞呢,线粒体基因的比例就会很高,我们将比例过高的细胞过滤掉即可。
我们使用以下命令,创建一个线粒体基因列表。
pbmc[[‘percent.mt’]] <- PercentageFeatureSet(pbmc,pattern = “^MT-“)
pattern = “^MT-“线粒体基因会有标注MT这里我们设置Patterns为MT即可。
VlnPlot(pbmc,features = c(“nFeature_RNA”,”nCount_RNA”,”percent.mt”),ncol=3)
选择三个指标”nFeature_RNA”,”nCount_RNA”,”percent.mt”绘制数据的小提琴图,
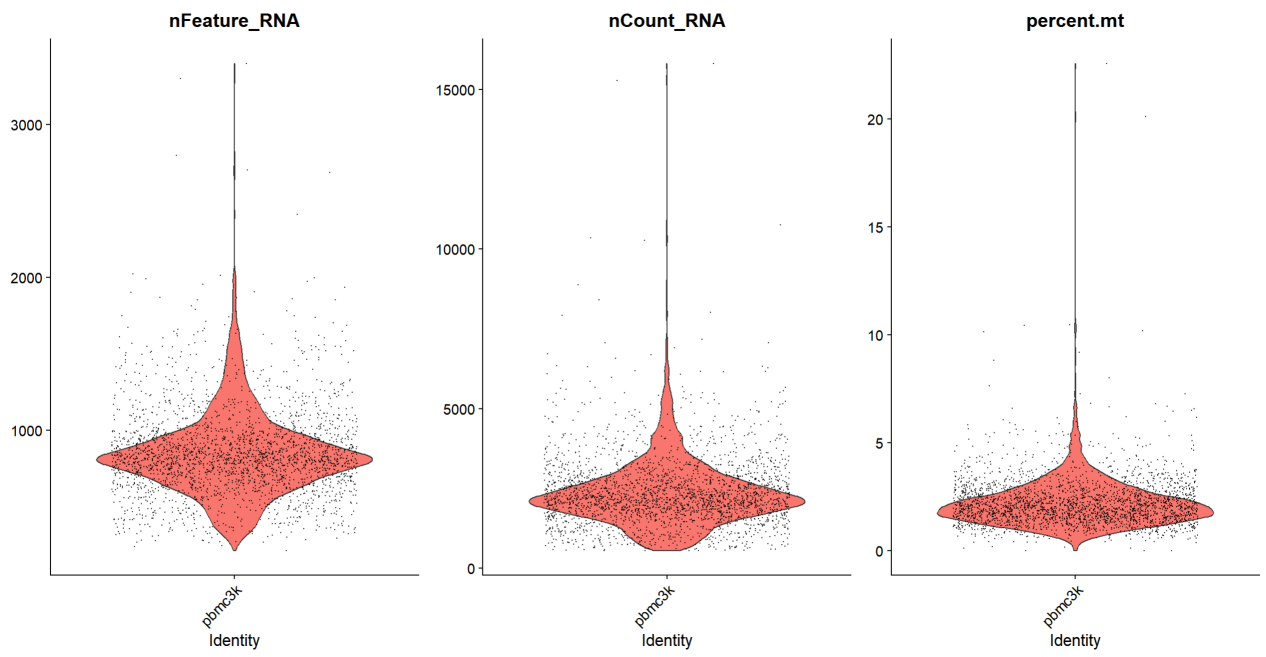
图中的每个点即为一个单细胞,可以看到有零星几个细胞处在图的上方,这些点会影响分析的结果,我们将其过滤掉。
过滤条件根据不同测序项目进行调整
pbmc <- subset(pbmc, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & percent.mt < 5)
这里我们过滤 feature counts> 2,500 or < 200,>5% mitochondrial counts
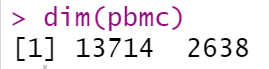
过滤后基因数目下降至13714个,细胞数目下降至2638
plot1 <- FeatureScatter(pbmc,feature1 = “nCount_RNA”,feature2 = “percent.mt”)
plot2 <- FeatureScatter(pbmc,feature1 = “nCount_RNA”,feature2 = “nFeature_RNA”)
plot1+plot2
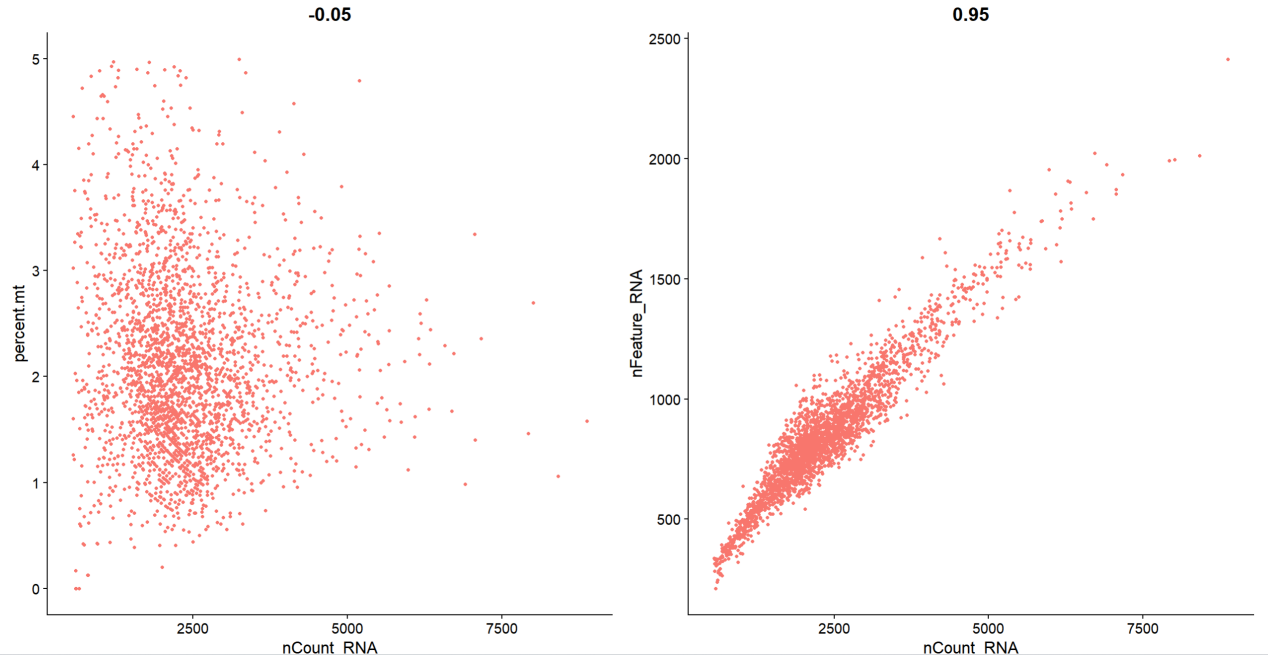
过滤后线粒体基因都处在5%以下,单细胞的数据随着umi counts数目增多,基因数目也随之增多,呈现线性关系。
标准化数据
由于每个细胞测序深度不同(有的细胞测的counts数目多,基因多,而有的细胞counts少基因少)我们需要对其进行标准化,才能进行进一步的差异表达计算。
pbmc <- NormalizeData(pbmc, normalization.method = “LogNormalize”, scale.factor = 10000)
这里我们选择常用的LogNormalize算法。scale.factor选择默认。
识别差异表达基因
#默认2000个,可以通过nfeatures调整
pbmc <- FindVariableFeatures(pbmc, selection.method = “vst”, nfeatures = 2000)
接下来我们可以使用FindVariableFeatures函数来进行差异基因识别。
top10 <- head(VariableFeatures(pbmc), 10)
前10基因
可视化前10基因
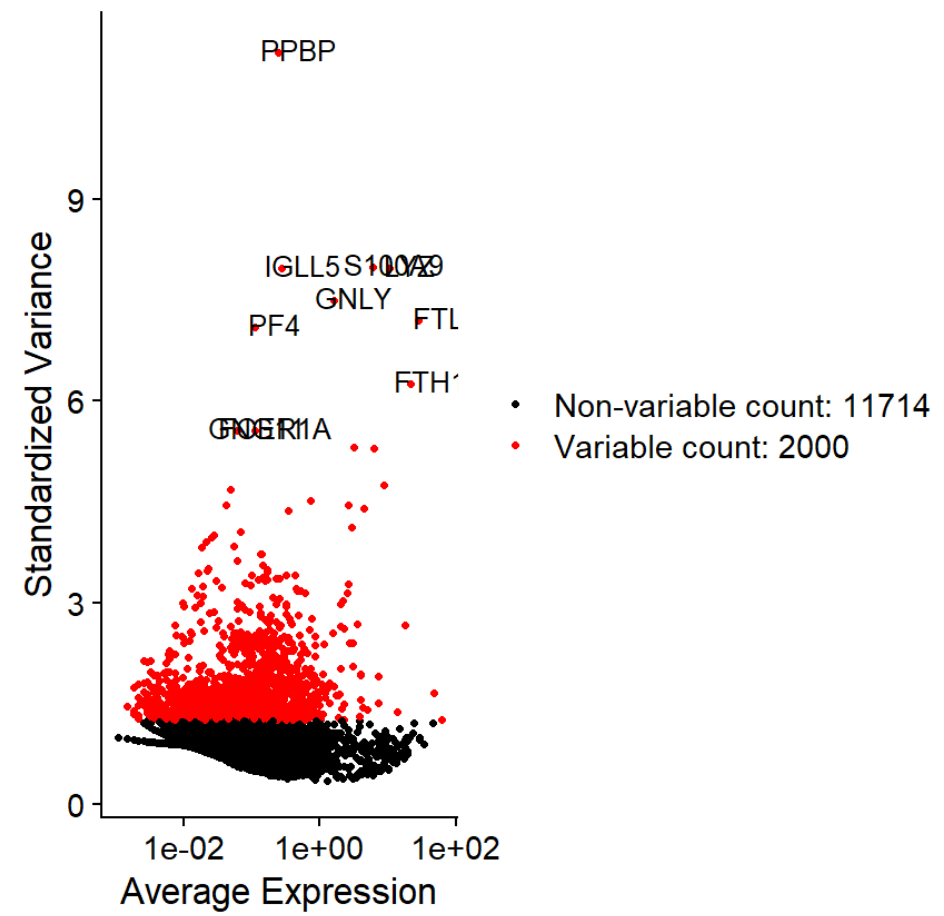
plot1 <- VariableFeaturePlot(pbmc)
plot1
plot2 <- LabelPoints(plot = plot1,points = top10)
plot2
plot1+plot2
预知后续的分析如何,请关注本公众号,小果带你领略生信分析的魅力。
单细胞测序分析内容欢迎尝试本公司新开发的云平台生物信息分析小工具,零代码完成分析,云平台网址:http://www.biocloudservice.com/home.html。