在统计假设检验中,testing-based procedures是一类常用的方法,用于在生物信息数据学中进行特征选择和变量选择。这些方法旨在从大量的生物信息数据中识别出最重要、最相关的特征或变量,以构建最佳的预测模型或解释生物学现象。
在生物信息学中,常常面对大量的基因表达数据、蛋白质序列数据或其他生物学特征数据。这些数据通常包含大量的变量,但并不是所有的变量都对所研究的生物学问题具有重要影响。因此,通过testing-based procedures,可以帮助筛选出那些与所关注生物学特性密切相关的变量,从而减少模型复杂性,提高预测和解释的准确性。
testing-based procedures中的一些常见方法包括backward selection(逐步后向选择)、forward selection(逐步前向选择)和stepwise regression(逐步回归)。逐步后向选择从包含所有预测变量的模型开始,通过逐步剔除那些对研究结果影响较小的变量,从而逐渐构建一个简化的模型。逐步前向选择则从一个空模型开始,逐步添加对目标变量有显著影响的变量。而逐步回归是将逐步前向选择和逐步后向选择结合在一起,每步考虑同时添加和删除变量。
这些方法通常会基于统计学指标(如p值、AIC、BIC等)来评估每个变量的重要性和显著性。通过反复添加或删除变量,最终得到一个较为简化的模型,其中包含最重要的预测变量,能够更好地解释和预测生物学现象。
1. 逐步后向选择(Backward Selection):
逐步后向选择是一种特征选择的方法,它从包含所有预测变量的完整模型开始,并通过逐步剔除对目标变量影响较小的变量来构建一个简化的模型。其步骤如下:
a. 首先,将所有预测变量包含在模型中。
b. 然后,通过一定的准则(如p值、AIC、BIC等)来检验每个变量的显著性,剔除其中对目标变量影响最不显著的一个。
c. 重复步骤b,直到剩下的变量都被认为是显著的或满足某个预设的停止准则。
示例:
我们将使用R中的”iris”数据集,这是一个包含了150个鸢尾花样本的数据集,其中每个样本有四个特征:花萼长度(Sepal.Length)、花萼宽度(Sepal.Width)、花瓣长度(Petal.Length)和花瓣宽度(Petal.Width)。我们将尝试使用逐步后向选择来建立一个回归模型,其中以花瓣宽度(Petal.Width)作为目标变量,其他特征作为预测变量。
# 加载必要的库
> library(stats)
# 加载数据集
> data(iris)
# 创建线性回归模型(完整模型),将Petal.Width作为目标变量,其他数值型变量作为预测变量
> full_model <- lm(Petal.Width ~ ., data = iris)
# 执行逐步后向选择
> backward_selection <- step(full_model, direction = “backward”)
Start: AIC=-531.74
Petal.Width ~ Sepal.Length + Sepal.Width + Petal.Length + Species
Df Sum of Sq RSS AIC
<none> 3.9976 -531.74
– Sepal.Length 1 0.12062 4.1182 -529.28
– Petal.Length 1 0.68266 4.6802 -510.09
– Sepal.Width 1 0.71403 4.7116 -509.09
– Species 2 1.38273 5.3803 -491.18
# 输出逐步后向选择结果
> summary(backward_selection)
Call:
lm(formula = Petal.Width ~ Sepal.Length + Sepal.Width + Petal.Length +
Species, data = iris)
Residuals:
Min 1Q Median 3Q Max
-0.59239 -0.08288 -0.01349 0.08773 0.45239
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.47314 0.17659 -2.679 0.00824 **
Sepal.Length -0.09293 0.04458 -2.084 0.03889 *
Sepal.Width 0.24220 0.04776 5.072 1.20e-06 ***
Petal.Length 0.24220 0.04884 4.959 1.97e-06 ***
Speciesversicolor 0.64811 0.12314 5.263 5.04e-07 ***
Speciesvirginica 1.04637 0.16548 6.323 3.03e-09 ***
—
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
Residual standard error: 0.1666 on 144 degrees of freedom
Multiple R-squared: 0.9538, Adjusted R-squared: 0.9522
F-statistic: 594.9 on 5 and 144 DF, p-value: < 2.2e-16
# 绘制回归诊断图
> par(mfrow=c(2,2)) # 设置多图绘制参数
> plot(backward_selection) # 绘制回归诊断图
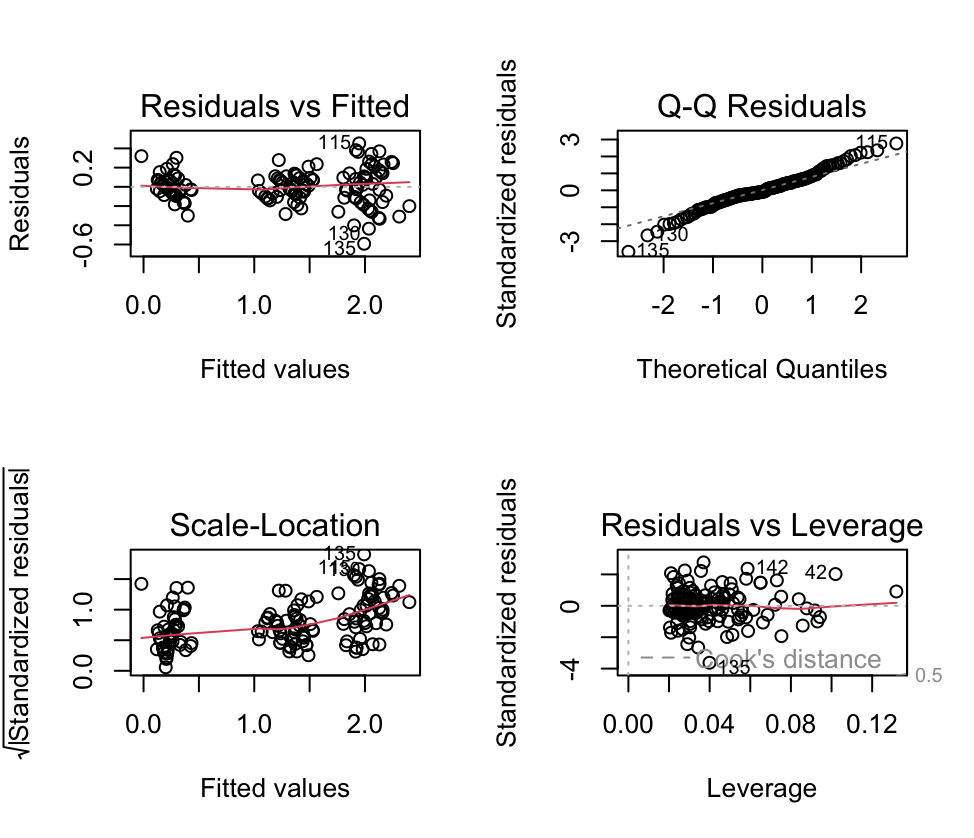
在上述代码中,我们加载了stats库并加载了”iris”数据集。然后,我们创建了一个包含所有数值型变量的线性回归模型(完整模型)。接着,通过step函数执行逐步后向选择,并将选择结果保存在backward_selection中。要可视化逐步后向选择的结果,我们可以使用plot函数绘制模型的诊断图。
这个示例演示了如何使用逐步后向选择方法来建立一个回归模型,使用基因表达数据集的特征作为预测变量,其中花瓣宽度(Petal.Width)是目标变量。您可以根据您的具体生物学数据替换目标变量和预测变量,以在实际应用中进行特征选择和回归建模。
2. 逐步前向选择(Forward Selection):
逐步前向选择是另一种特征选择的方法,它与逐步后向选择相反,从包含零个预测变量的空模型开始,逐步添加对目标变量影响显著的变量。其步骤如下:
a. 首先,将一个空模型作为起始点。
b. 然后,逐个将每个预测变量添加到模型中,并使用一定的准则(如p值、AIC、BIC等)来检验该变量的显著性。
c. 重复步骤b,每次添加对目标变量影响最显著的一个变量,直到进一步添加变量不再显著或满足某个预设的停止准则。
示例:
我们需要加载数据集并执行逐步前向选择。在这个示例中,我们将使用”diagnosis”作为目标变量(响应变量),其他数值型变量作为预测变量。
# 加载必要的库
> library(stats)
> library(caret)
# 加载数据集
> data(breast_cancer)
# 创建目标变量和预测变量的数据框
> data_df <- data.frame(diagnosis = breast_cancer$diagnosis,
breast_cancer[, 3:12]) # 选择3到12列的数据作为预测变量
# 将目标变量转换为二进制因子(因为我们要进行二元分类)
> data_df$diagnosis <- factor(data_df$diagnosis, levels = c(“B”, “M”),
labels = c(“Benign”, “Malignant”))
# 使用逐步前向选择构建线性回归模型
> forward_selection <- train(diagnosis ~ ., data = data_df,
method = “lmStepAIC”, direction = “forward”)
# 输出逐步前向选择结果
> summary(forward_selection$finalModel)
# 绘制拟合图
> plot(forward_selection$finalModel)
# 绘制显著性图
> par(mfrow=c(1,1))
> stepAIC::plot(forward_selection$finalModel)
在上述代码中,我们首先加载了stats和caret库,并加载了”breast_cancer”数据集。然后,我们创建了一个包含目标变量和预测变量的数据框”data_df”。接着,我们将目标变量”diagnosis”转换为二进制因子,以便进行二元分类。使用train函数,我们执行了逐步前向选择,并将选择结果保存在forward_selection中。在这里,我们使用lmStepAIC作为方法来进行逐步前向选择,并指定方向为”forward”,表示逐步添加显著的变量。要可视化逐步前向选择的结果,我们可以绘制模型的拟合情况,以及变量的显著性。
3. 逐步回归(Stepwise Regression):
逐步回归是将逐步前向选择和逐步后向选择结合在一起的方法。它通过在每个步骤中同时考虑添加和删除变量,来构建最优的模型。其步骤如下:
a. 首先,将一个空模型作为起始点。
b. 然后,按照一定的准则(如p值、AIC、BIC等)考虑添加和删除变量。
c. 在每个步骤中,可以同时添加一个变量和删除一个不显著的变量。
d. 重复步骤b和c,直到进一步添加和删除变量都不再显著或满足某个预设的停止准则。
示例:
我们需要加载“mtcars”数据集并执行逐步回归,在这个示例中,我们将使用“mpg”作为目标变量(响应变量),其他数值型变量作为预测变量。
# 加载必要的库
> library(stats)
# 加载数据集
> data(mtcars)
# 创建目标变量和预测变量的数据框
> data_df <- data.frame(mpg = mtcars$mpg,
+ mtcars[, c(2:4, 6:7)]) # 选择第2至4列和第6至7列的数据作为预测变量
# 使用逐步回归构建线性回归模型
> stepwise_model <- step(lm(mpg ~ ., data = data_df), direction = “both”)
Start: AIC=65.19
mpg ~ cyl + disp + hp + wt + qsec
Df Sum of Sq RSS AIC
– qsec 1 1.759 170.44 63.526
– disp 1 6.534 175.22 64.410
– hp 1 6.983 175.67 64.492
<none> 168.69 65.194
– cyl 1 16.950 185.63 66.258
– wt 1 73.848 242.53 74.813
Step: AIC=63.53
mpg ~ cyl + disp + hp + wt
Df Sum of Sq RSS AIC
– disp 1 6.176 176.62 62.665
<none> 170.44 63.526
– hp 1 18.048 188.49 64.746
+ qsec 1 1.759 168.69 65.194
– cyl 1 24.546 194.99 65.831
– wt 1 90.925 261.37 75.206
Step: AIC=62.66
mpg ~ cyl + hp + wt
Df Sum of Sq RSS AIC
<none> 176.62 62.665
– hp 1 14.551 191.17 63.198
+ disp 1 6.176 170.44 63.526
– cyl 1 18.427 195.05 63.840
+ qsec 1 1.401 175.22 64.410
– wt 1 115.354 291.98 76.750
# 输出逐步回归结果
> summary(stepwise_model)
Call:
lm(formula = mpg ~ cyl + hp + wt, data = data_df)
Residuals:
Min 1Q Median 3Q Max
-3.9290 -1.5598 -0.5311 1.1850 5.8986
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 38.75179 1.78686 21.687 < 2e-16 ***
cyl -0.94162 0.55092 -1.709 0.098480 .
hp -0.01804 0.01188 -1.519 0.140015
wt -3.16697 0.74058 -4.276 0.000199 ***
—
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
Residual standard error: 2.512 on 28 degrees of freedom
Multiple R-squared: 0.8431, Adjusted R-squared: 0.8263
F-statistic: 50.17 on 3 and 28 DF, p-value: 2.184e-11
# 绘制回归诊断图
> par(mfrow=c(2,2)) # 设置多图绘制参数
> plot(stepwise_model) # 绘制回归诊断图
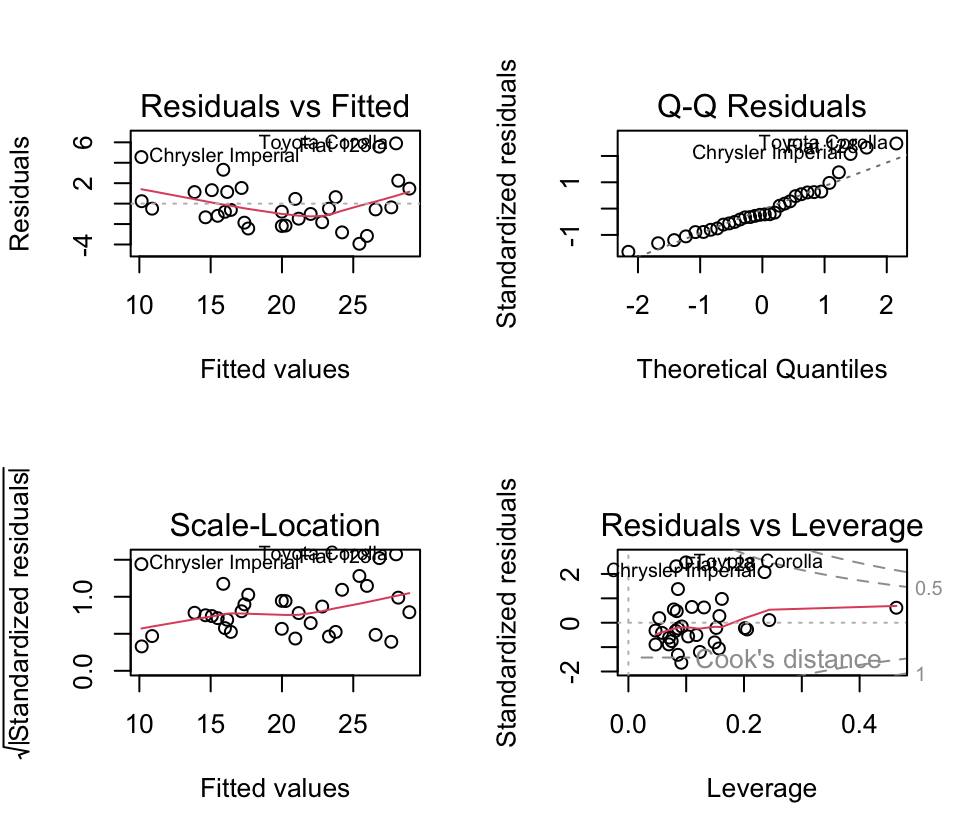
在上述代码中,我们加载了stats库,并加载了”mtcars”数据集。然后,我们创建了一个包含目标变量和预测变量的数据框”data_df”。接着,我们使用lm函数创建了一个线性回归模型(完整模型)。然后,通过step函数执行逐步回归,并将选择结果保存在stepwise_model中。要可视化逐步回归的结果,我们可以使用plot函数绘制模型的诊断图。
这个示例演示了如何使用逐步回归方法来建立一个回归模型,使用与生物学相关的数据集特征作为预测变量,其中”mpg”是目标变量。
以上就是对假设测试检验对过程的简单介绍啦,需要注意的是,虽然testing-based procedures是一种常用的特征选择方法,但在应用时需要谨慎。过度使用这些方法可能会导致过拟合问题,即模型在训练数据上表现良好,但在未知数据上表现较差。因此,研究人员需要在使用这些方法时进行适当的验证和交叉验证,以确保所选取的变量在不同数据集上都能够稳定地发挥作用,并得到可靠的生物学解释。
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小果哦~~或者也可以关注我们的官网也会持续更新的哦~ http://www.biocloudservice.com/home.html
References: