大家好,小果今天继续介绍R包Seurat单细胞测序分析。
上一期我们用Seurat对数据进行了进一步的过滤,过滤掉了不好的细胞(死细胞,空细胞,多细胞),提高了数据的质量,并对数据进行了标准化。今天我们将使用不同的算法来对数据进行降维。
为什么要做降维呢?
人类细胞计划根据基因表达模式不同来分类细胞。有的细胞分化后,只会表达特定的基因,不会再表达其他的基因了,根据表达的基因来分类细胞是可靠的。
在小果案例这个数据集中有2700个细胞,我们不可能一个一个去看,它们当中有很多表达模式是相似的,我们可以根据基因表达模式不同来进行分类。
三种常用降维算法
PCA主成分分析
其原理是通过对协方差矩阵进行特征分解,以得出数据的主成分与它们的权值,即特征值和特征向量。它产生的结果可以理解为对原数据中的方差做出解释:哪一个方向上的数据值对方差的影响最大?
通俗点说,就像选人大代表一样。每个阶层,每个民族,每个职业选代表。 在单细胞分析中就是对细胞分组,每组选代表性的基因选出来。
t-SNE
t-SNE是一种流行的探索高维数据的方法,由2008年,范德马顿和辛顿发明。它具有近乎神奇的能力,可以从具有数百甚至数千维的数据中创建引人注目的两维“图片”。想继续深入理解的小伙伴可以去下面这个网址学习t-SNE。
How to Use t-SNE Effectively (distill.pub)
UMAP
UMAP是McInnes等人2019年发明的一项新的数据降维技术。与t-SNE相比,它具有许多优势,最显着的是提高了速度并更好地保存了数据的全局结构。感兴趣的小伙伴可以去下面这个网址交互式理解UMAP的降维原理。
Understanding UMAP (pair-code.github.io)
用Seurat对单细胞测序数据降维
我们对这2700个细胞进行分类,首先假设分成10组。
#将数据所有基因名保存到all.genes变量中。
all.genes <- rownames(pbmc)
#默认处理2000个基因,添加features对所有基因进行处理
pbmc <- ScaleData(pbmc,features = all.genes)
#如果需要移除不想要的基因,可以添加vars.to.regress
pbmc <- ScaleData(pbmc, vars.to.regress = “percent.mt”)
这里移除所有的线粒体基因。
#PCA分析与可视化
pbmc <- RunPCA(pbmc,features = VariableFeatures(object = pbmc))
使用RunPCA函数对数据pbmc进行PCA分析。
#输出前五个PCA结果
print(pbmc[[‘pca’]],dims = 1:9,nfeatures = 5)

#PCA可视化,Seurat提供三个函数VizDimReduction(), DimPlot(), DimHeatmap()
VizDimLoadings(pbmc,dims = 1:2,reduction = “pca”)
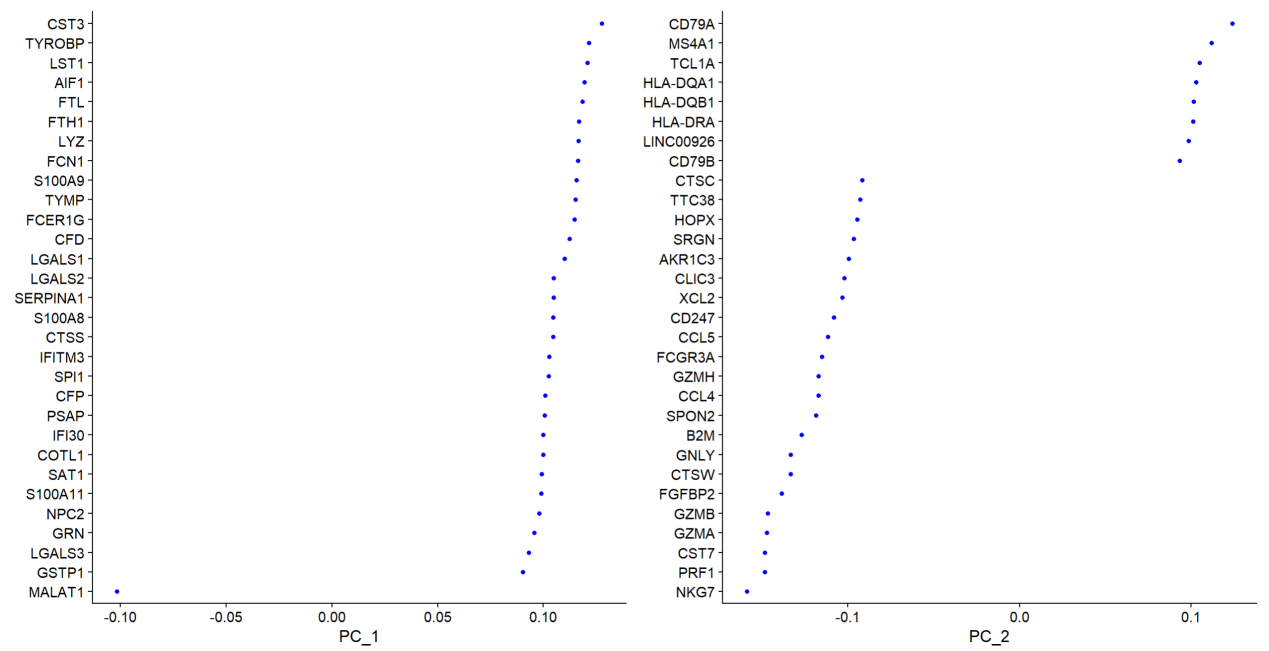
Pca降维主成分1和2的差异显著基因
DimPlot(pbmc,reduction = “pca” )
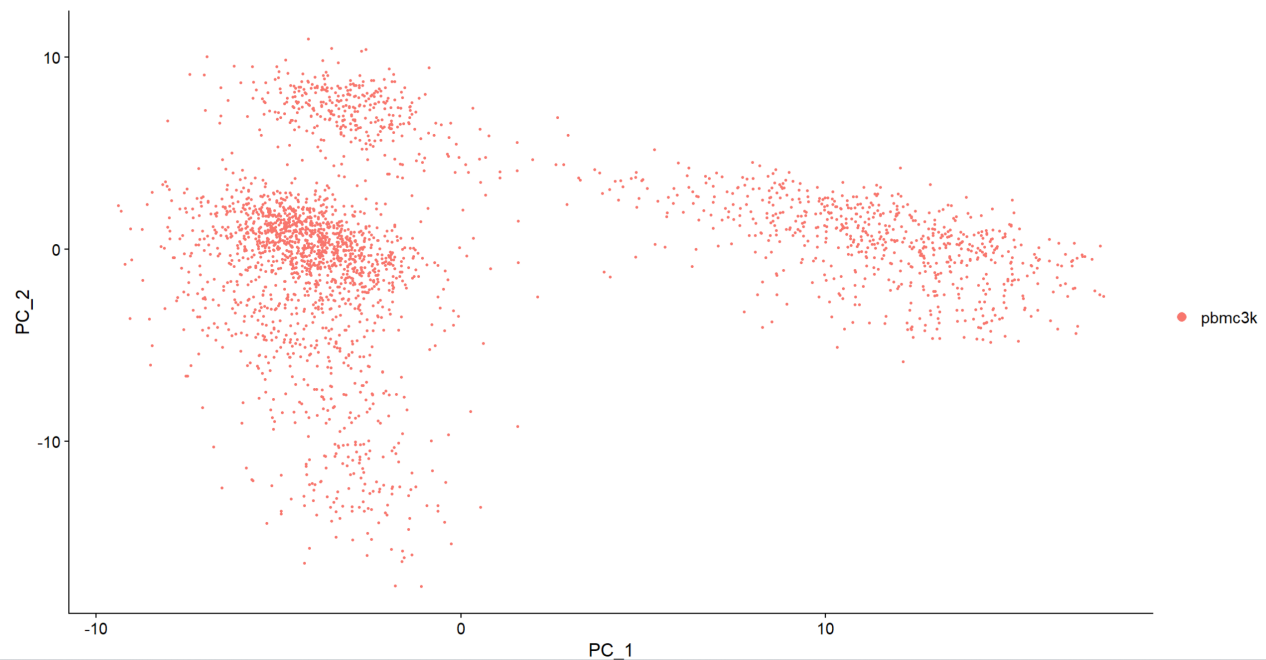
使用PCA方法对细胞分类。
#绘制10个
DimHeatmap(pbmc, dims = 1:10, cells = 500, balanced = TRUE)

可以看到,到了后面已经不能很好的将细胞区分开了。
确定分组个数
#确定数据的分群个数,比较耗时
pbmc <- JackStraw(pbmc,num.replicate = 100)
pbmc <- ScoreJackStraw(pbmc,dims = 1:20)
#绘制碎石图,确定分群个数
JackStrawPlot(pbmc,dims = 1:20)
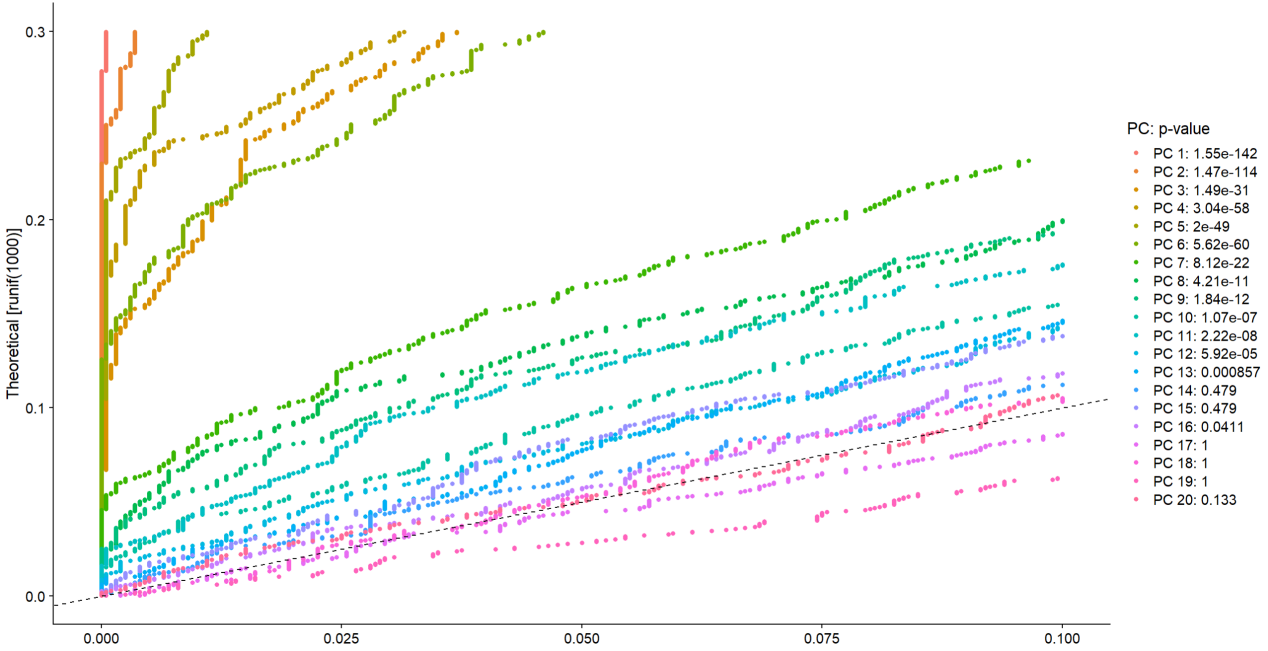
虚线以上是算法建议分成的组数。
ElbowPlot(pbmc)
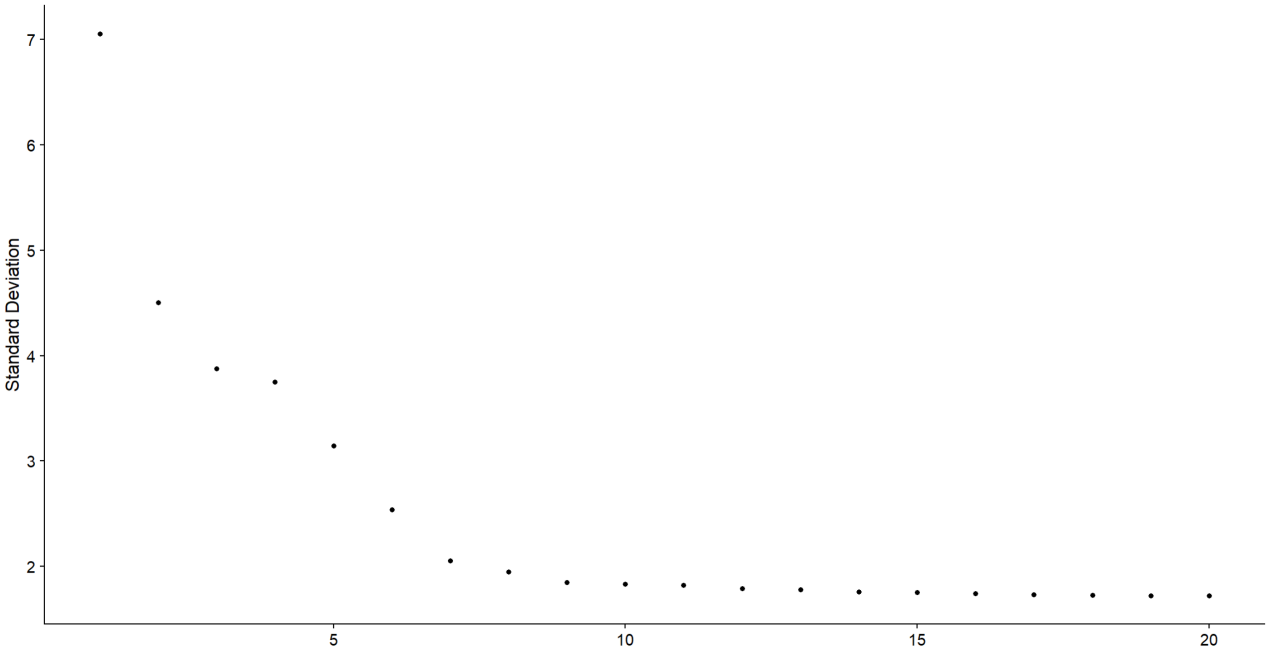
从图中可以看出,10以后没有什么变化了,说明推荐分成10组。
细胞聚类
pbmc <- FindNeighbors(pbmc,dims = 1:10)
FindNeighbors函数计算细胞间距离,如果两个细胞表达谱越相近,距离越小
pbmc <- FindClusters(pbmc,resolution = 0.5)
#查看不同细胞的分群
head(Idents(pbmc),5)
查看前5个群
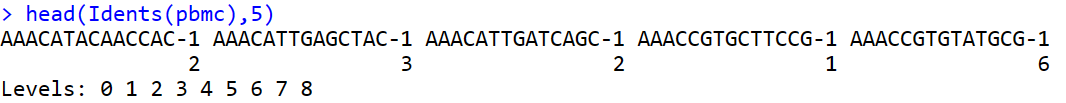
虽然我们设置dims=10,分成10组,但是通过聚类后观察LevelsLevels说明细胞被分成了9组。

barplot(table(Idents(pbmc)))
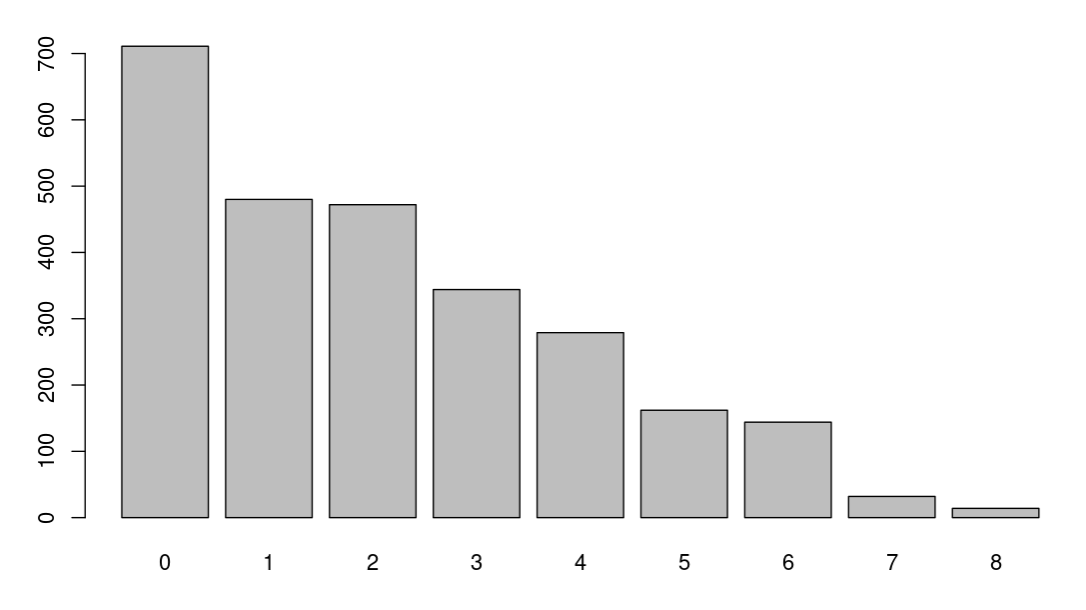
列出了每个群的细胞数目
t-SNE细胞聚类与可视化
pbmc <- RunTSNE(pbmc,dims = 1:10)
p2 <- DimPlot(pbmc,reduction = “tsne”)
p2
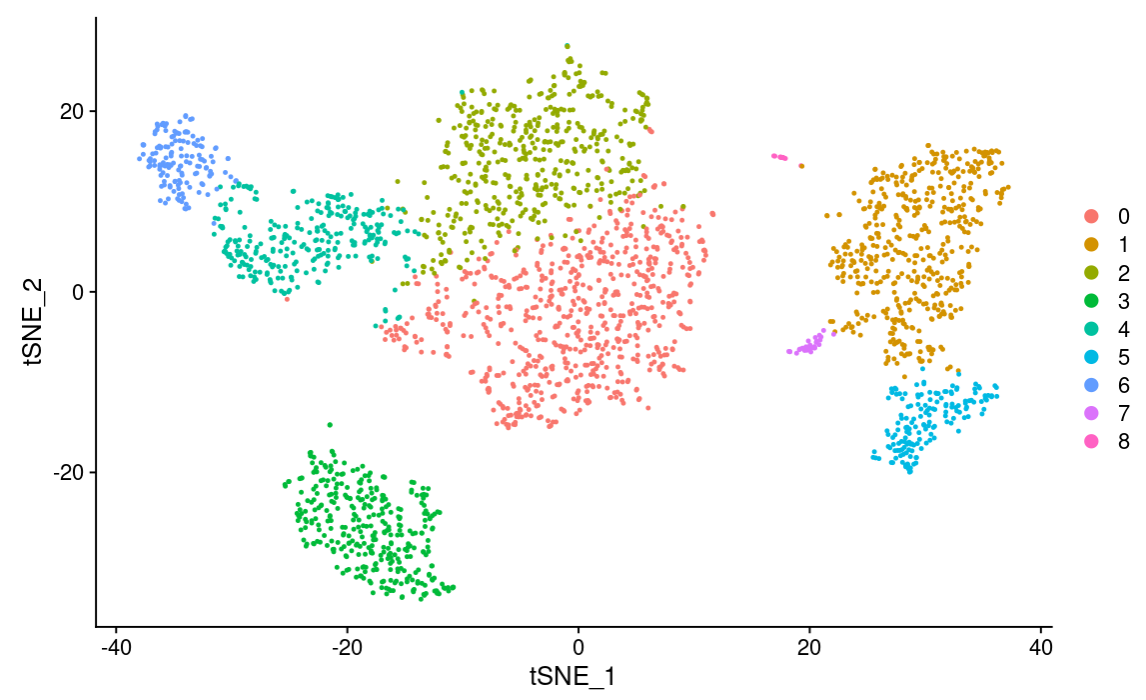
T-SNE分类结果图。
UMAP细胞聚类与可视化
pbmc <- RunUMAP(pbmc,dims = 1:10,label=T)

Reductions存储着降维的结果,第一个是pca分析的结果,umap随后被追加入了进去。
我们可以将计算好的UMAP结果输出为单独的文件,用loupe browse查看。
#绘制UMAP图
p1 <- DimPlot(pbmc,reduction = “umap”)
p1
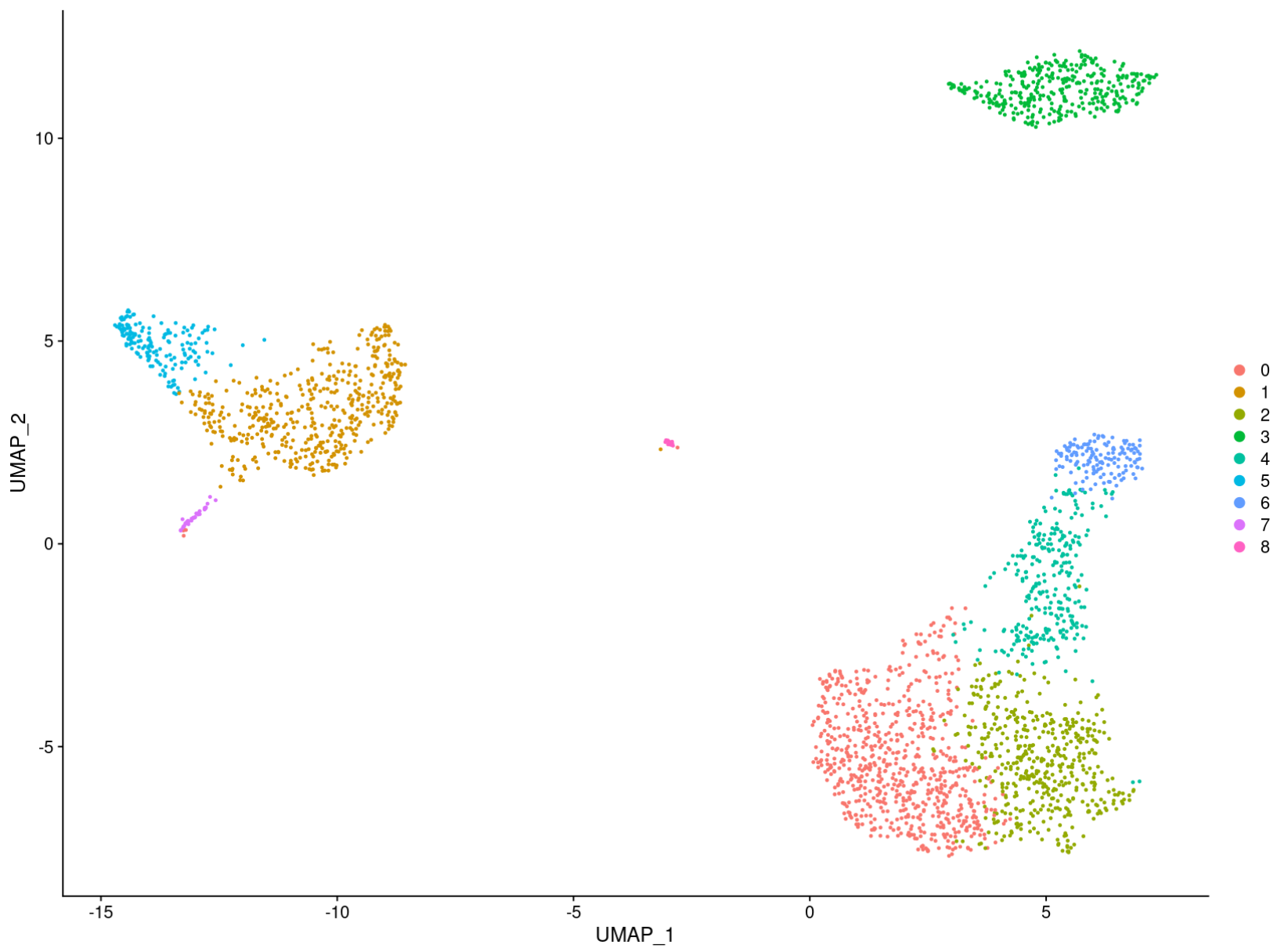
UMAP分类结果图。
UMAP、t-SNE与PCA对比
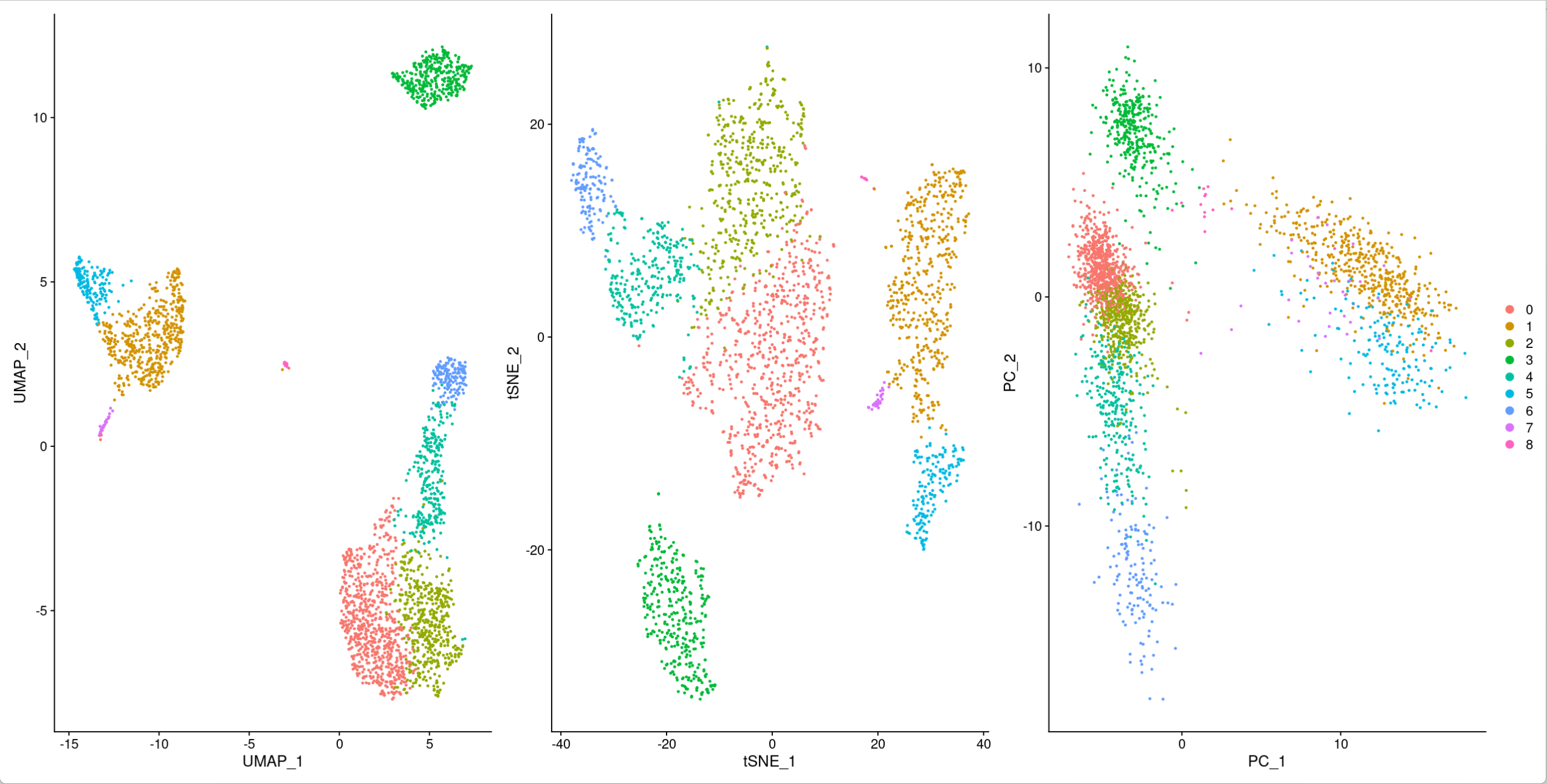
PCA分类效果最差,明显没有分开。UMAP较t-SNE分类效果更好。
好您是否明白了呢,如果有不懂的来和小果讨论吧~下期小果将对分类的细胞进行鉴定与寻找标记基因。我们不见不散~