我们大家知道基因与多种免疫细胞类型之间有着相关性,那么了解指定基因与免疫细胞类型之间的相互作用,以及是否存在相关性,以及基因在调节免疫反应中的潜在功能是很重要的。那么下面我们先了解一下r包的概念。
CIBERSORT是一种常用于解析肿瘤组织中不同免疫细胞类型分布的算法。通过将CIBERSORT结果与特定基因的表达数据进行相关性分析,可以揭示这些免疫细胞类型在基因调控过程中的潜在重要性。
本期主要给大家介绍单基因与多种免疫细胞的相关性分析的实操保姆级代码。
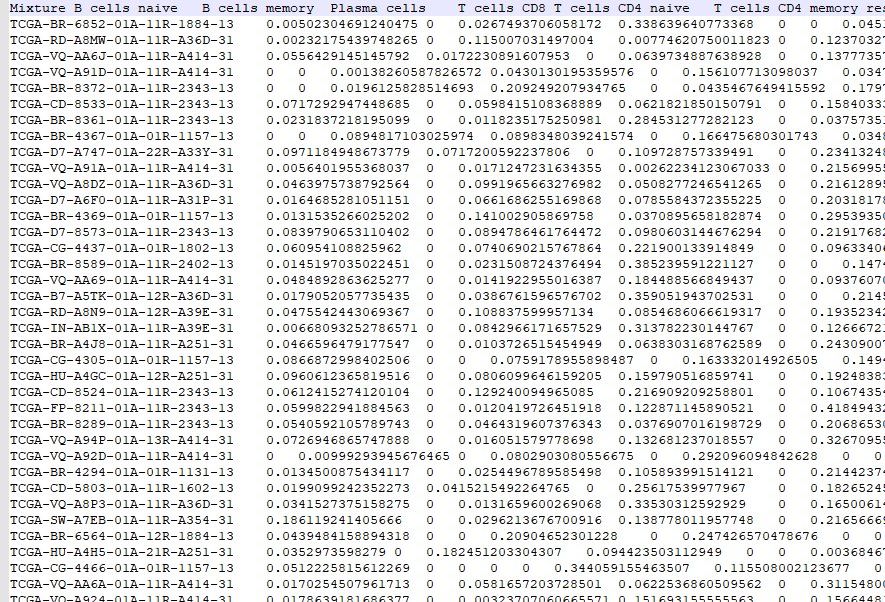
代码中对示例基因的CXCR4的表达量进行免疫相关分析。下面是下载整理的样本表达量信息和样本免疫细胞信息。

#if (!requireNamespace(“BiocManager”, quietly = TRUE))
# install.packages(“BiocManager”)
#BiocManager::install(“limma”)
#检查是否安装了“BiocManager”包。如果没有安装,它会进行安装。BiocManager用于在R中管理Bioconductor包。
#install.packages(“ggplot2”)
#install.packages(“ggpubr”)
#install.packages(“ggExtra”)
library(limma)
library(ggplot2)
library(ggpubr)
library(ggExtra)
#安装和加载所需的R包,例如“limma”、“ggplot2”、“ggpubr”和“ggExtra”。这些包用于各种数据分析和可视化任务。
immuneFile=”CIBERSORT-Results.txt” #免疫结果文件
expFile=”symbol.txt” #表达输入文件
gene=”CXCR4″ #基因名称
pFilter=0.05 #CIBERSORT结果过滤条件
#如“immuneFile”(包含CIBERSORT结果的文件)、
#“expFile”(表达式输入文件)、“gene”(感兴趣的特定基因)和“pFilter”(CIBERSORT结果过滤阈值)。
#读取免疫结果文件,并对数据进行整理
immune=read.table(immuneFile,sep=”\t”,header=T,row.names=1,check.names=F)
immune=immune[immune[,”P-value”]<pFilter,]
immune=as.matrix(immune[,1:(ncol(immune)-3)])
#从“immuneFile”中读取CIBERSORT结果,
#并过滤掉p值小于指定阈值(“pFilter”)的行。还从“immune”数据框中删除不必要的列。
#读取表达文件,并对输入文件整理
rt=read.table(expFile,sep=”\t”,header=T,check.names=F)
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)
#从“expFile”中读取基因表达数据,提取指定“gene”的相关数据,
#并处理数据以去除正常样本并选择与免疫数据交集的样本。
#然后,选择除第一列外的所有列作为表达数据,并重新定义维度名称。最后,数据进行了平均处理。
group=sapply(strsplit(colnames(data),”\\-“),”[“,4)
group=sapply(strsplit(group,””),”[“,1)
group=gsub(“2″,”1”,group)
data=data[,group==0]
#去除正常样品,根据样本名中的信息判断是否为正常样本,并去除了正常样本,保留仅包含肿瘤样本的数据。
#样品取交集将基因的表达值添加到免疫数据中,然后取免疫数据和基因表达数据的交集,
#以确保两者具有相同的样本。这样可以保证免疫数据和基因表达数据之间有相同的样本,便于后续相关性分析。
data=rbind(data,gene=data[gene,])
exp=t(data[c(“gene”,gene),])
sameSample=intersect(row.names(immune),row.names(exp))
immune1=immune[sameSample,]
exp1=exp[sameSample,]
outTab=data.frame()
x=as.numeric(exp1[,1])
#按免疫细胞循环
for(j in colnames(immune1)[1:22]){
y=as.numeric(immune1[,j])
if(sd(y)>0.001){
df1=as.data.frame(cbind(x,y))
corT=cor.test(x,y,method=”spearman”)
cor=corT$estimate
pValue=corT$p.value
p1=ggplot(df1, aes(x, y)) +
ylab(j)+xlab(gene)+
geom_point()+ geom_smooth(method=”lm”,formula=y~x) + theme_bw()+
stat_cor(method = ‘spearman’, aes(x =x, y =y))
if(pValue<pFilter){
pdf(file=paste0(j,”.pdf”),width=5,height=5)
print(p1)
dev.off()
outTab=rbind(outTab,cbind(Cell=j,pValue))
}
}
}
#相关性检验进行相关性检验,计算基因表达值(x)与各种免疫细胞类型比例(y)之间的Spearman秩相关系数。
#循环遍历每种免疫细胞类型,并绘制散点图和回归线,以及标注相关系数和p值。
#如果相关性显著(pValue<pFilter),则将散点图保存为以免疫细胞类型命名的PDF文件,
#并将结果存储在数据框”outTab”中。最后,将包含显著相关性的结果存储在名为”immuneCor.result.txt”的文本文件中
write.table(outTab,file=”immuneCor.result.txt”,sep=”\t”,row.names=F,quote=F)
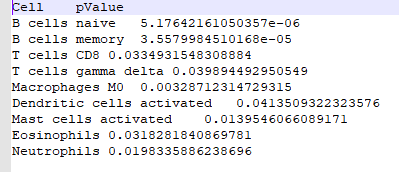
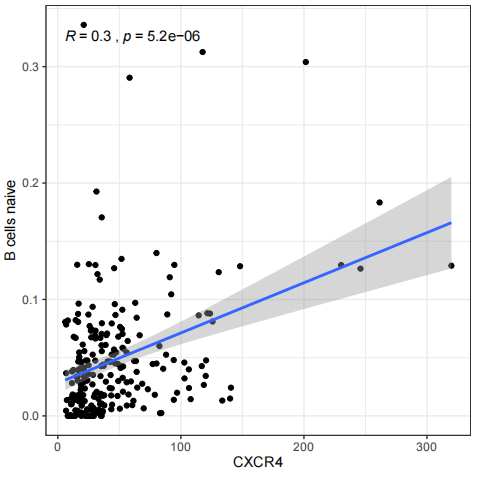
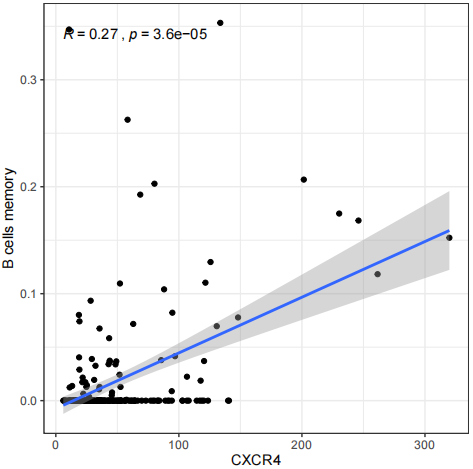
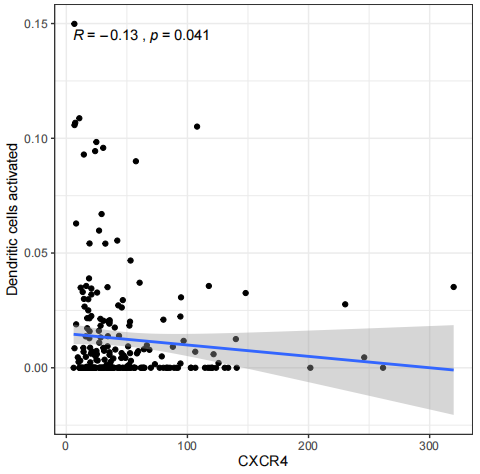
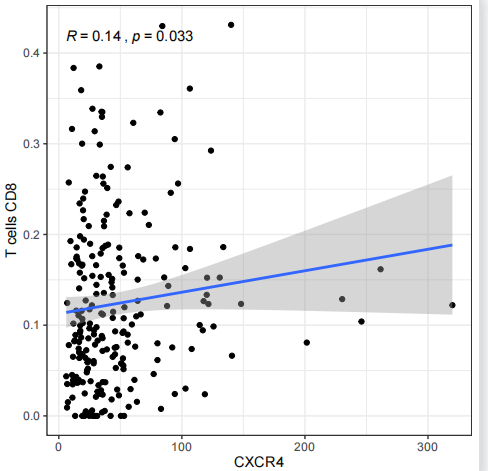
总之,在免疫与基因表达之间寻找关联性、了解肿瘤免疫微环境并发现新的生物学信息具有重要意义。下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:http://www.biocloudservice.com/home.html。
云生信平台链接:http://www.biocloudservice.com/home.html。