要谈论到生物信息学就业最热门的领域,药物分子设计绝对是绕不开的一大话题。可能很多生信的小伙伴们认为生信的主要工作领域在各种数据的处理和挖掘中,从而对药物分子设计这一领域有些陌生。从今天开始,小果就给大家带来一系列详细的药物分子设计流程教学。废话不多说,我们直接开始吧!
什么是Discovery Studio?
Discovery Studio是由DNASTAR公司开发的一套集成的计算化学和分子建模软件套件。它被广泛用于药物研发、蛋白质研究、生物分子模拟等领域。
Discovery Studio具有丰富的功能和工具,可用于药物设计、虚拟筛选、蛋白质结构预测、分子动力学模拟等任务。
下面小果给大家带来了Discovery Studio的一些常见的功能和模块介绍:
- LigandFit(分子对接和融合):该模块可用于药物设计和虚拟筛选。它允许研究人员将小分子(药物候选物)与蛋白质靶点进行对接,以预测它们之间的相互作用和结合能力。此外,LigandFit还支持分子融合技术,用于在已知配体的基础上生成新的分子。
- Sequence Viewer(蛋白质序列分析和比较):该模块可用于分析和比较蛋白质序列。它提供了多个工具,包括多序列比对、蛋白质结构预测和功能注释。通过Sequence Viewer,研究人员可以确定蛋白质序列之间的共同特征,并推断它们之间的结构和功能关系。
- QSAR模块(定量构效关系):该模块主要用于构建和预测定量构效关系模型。借助QSAR模块,研究人员可以根据分子的结构和活性数据进行模型训练,并使用此模型预测新化合物的生物活性。这有助于加快药物研发过程,筛选出具有潜在药物活性的化合物。
- CHARMm(分子动力学模拟):该模块可用于进行分子动力学模拟,模拟蛋白质、核酸和小分子在水溶液中的动态行为。研究人员可以通过CHARMm模块了解分子的构象演变、相互作用以及其与环境之间的影响。这对于理解蛋白质功能和药物-靶点相互作用机制至关重要。
- GCG(生物序列分析):GCG模块是一个强大的工具,用于分析和处理生物序列数据,包括DNA、RNA和蛋白质序列。它包括多个计算工具,如比对工具、搜索工具和注释工具,方便了研究人员对生物序列进行精确的分析和解释。
通过上面几个功能的介绍,小果相信小伙伴们已经初步理解了Discovery Studio的强大之处,是不是已经迫不及待想知道具体如何使用它了呢?现在就跟上小果的节奏,迅速走近它!
绘制小分子配体
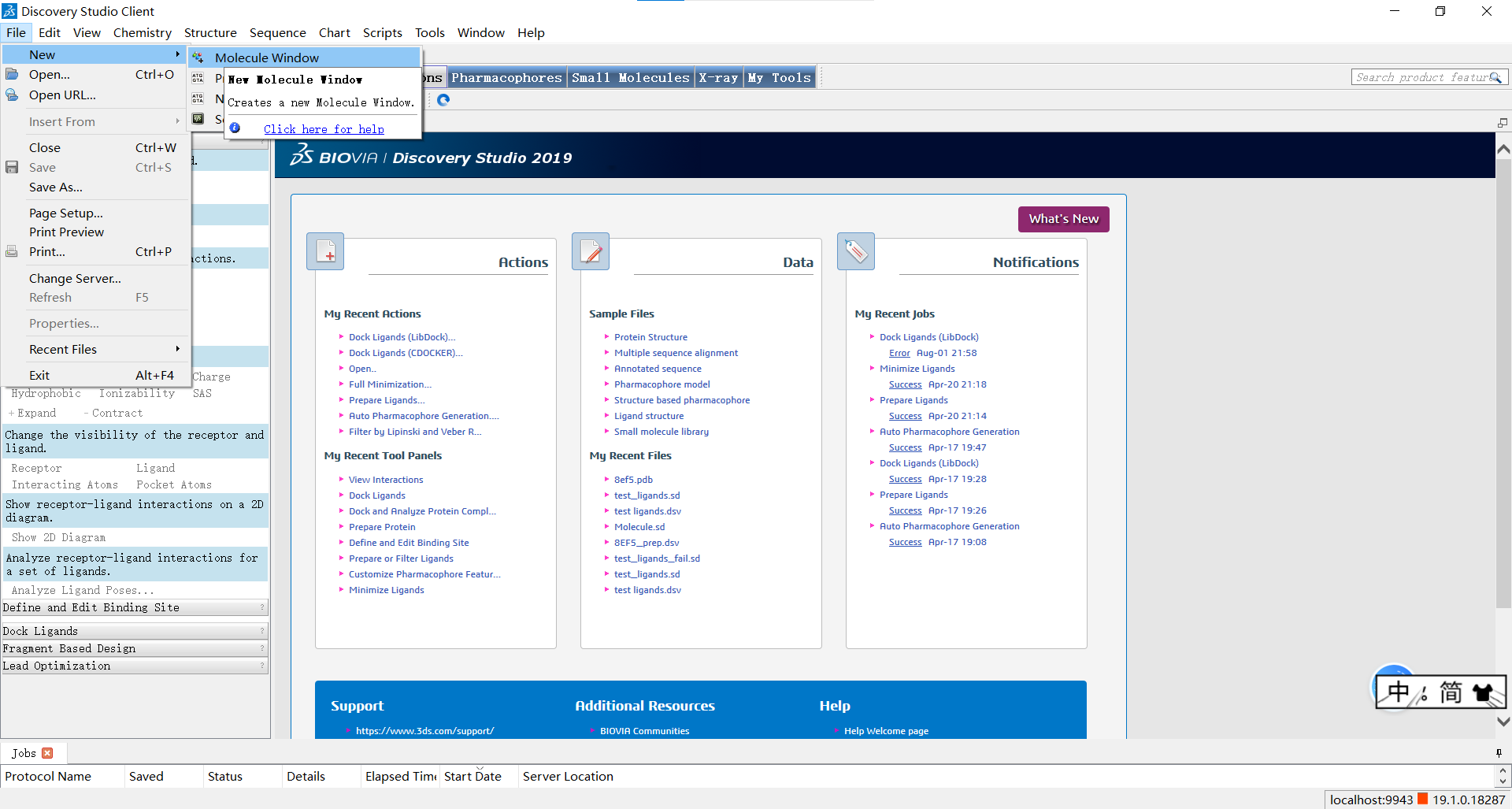
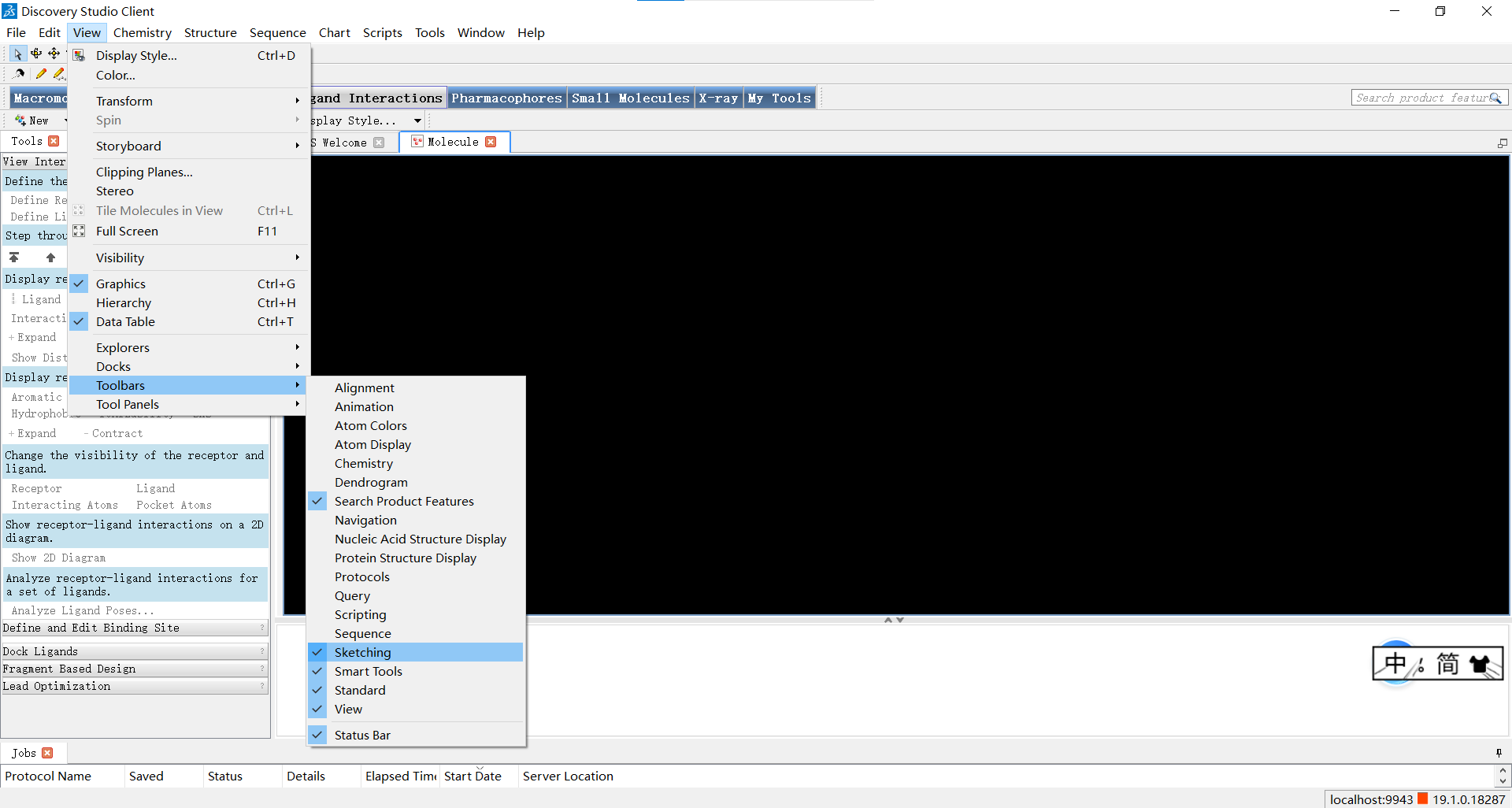
如图所示,小果先新建了一个小分子窗口,随后再view->toolbar中勾选sketching和view两个选项,这样我们就可以通过左上角的工具栏按我们的心意创建小分子啦

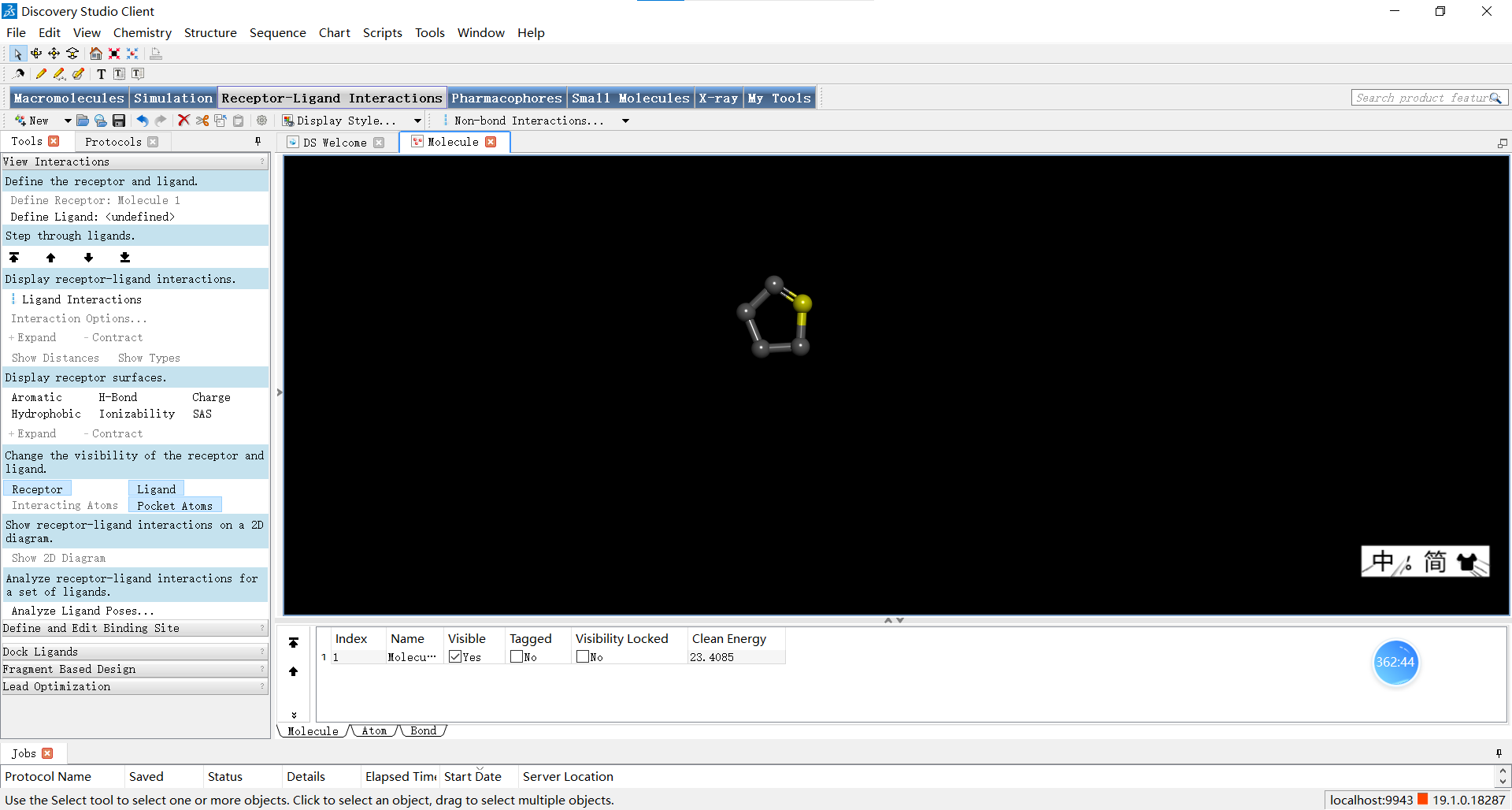
如图小果标出的铅笔按钮可以用来绘制原子和化学键,小果在这里画了一个五元环为例,给大家展示一下绘制小分子基本框架的方法。
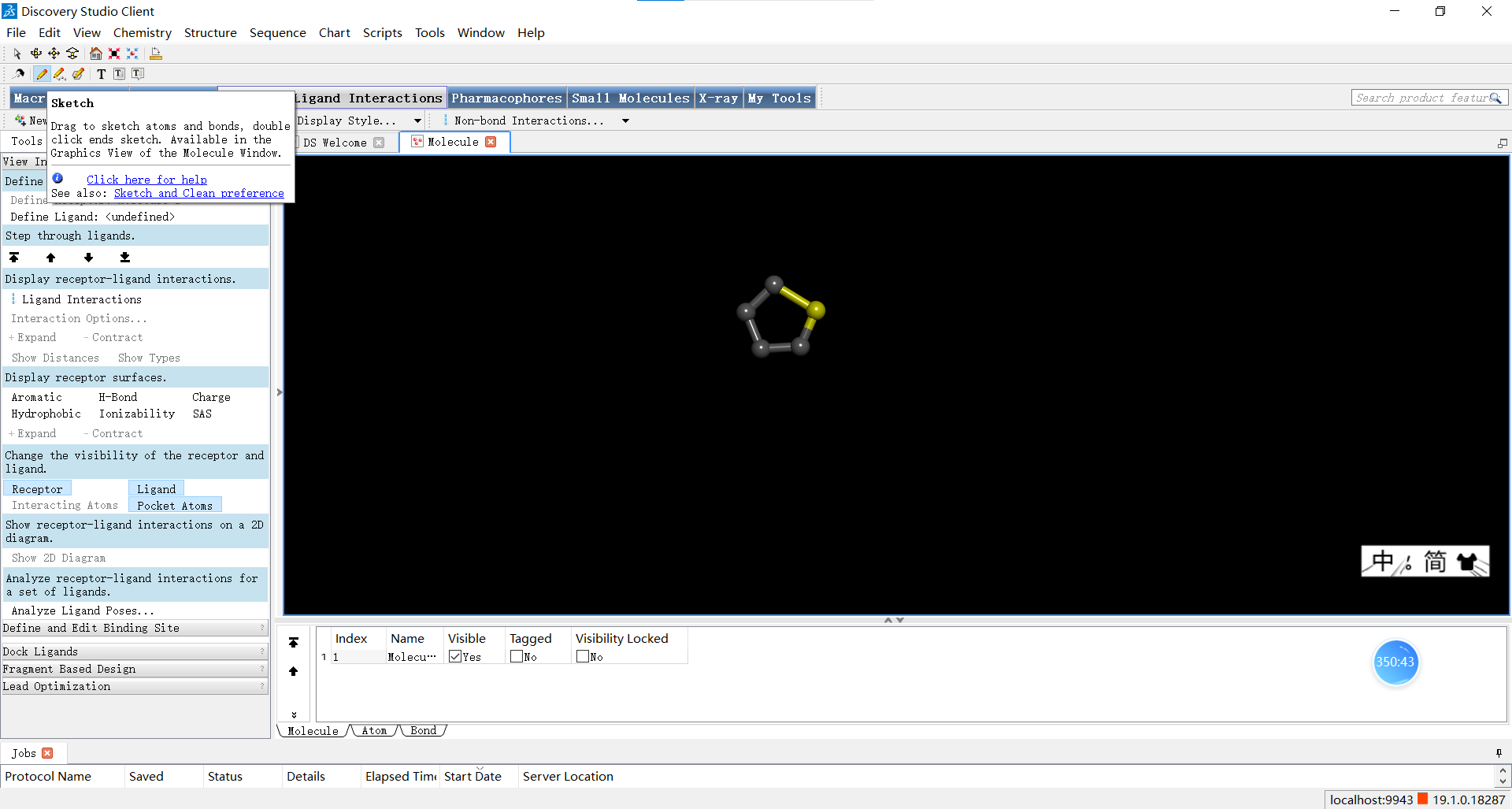
如果我们想改变化学键,可以先在左上角切换至箭头图标来选择想要改变的键,随后鼠标右击打开菜单,在bond菜单中对化学键的类型进行调整。在这个右键拉开的菜单中我们还可以进行改变某个键或原子显示颜色或添加备注等等其他操作。
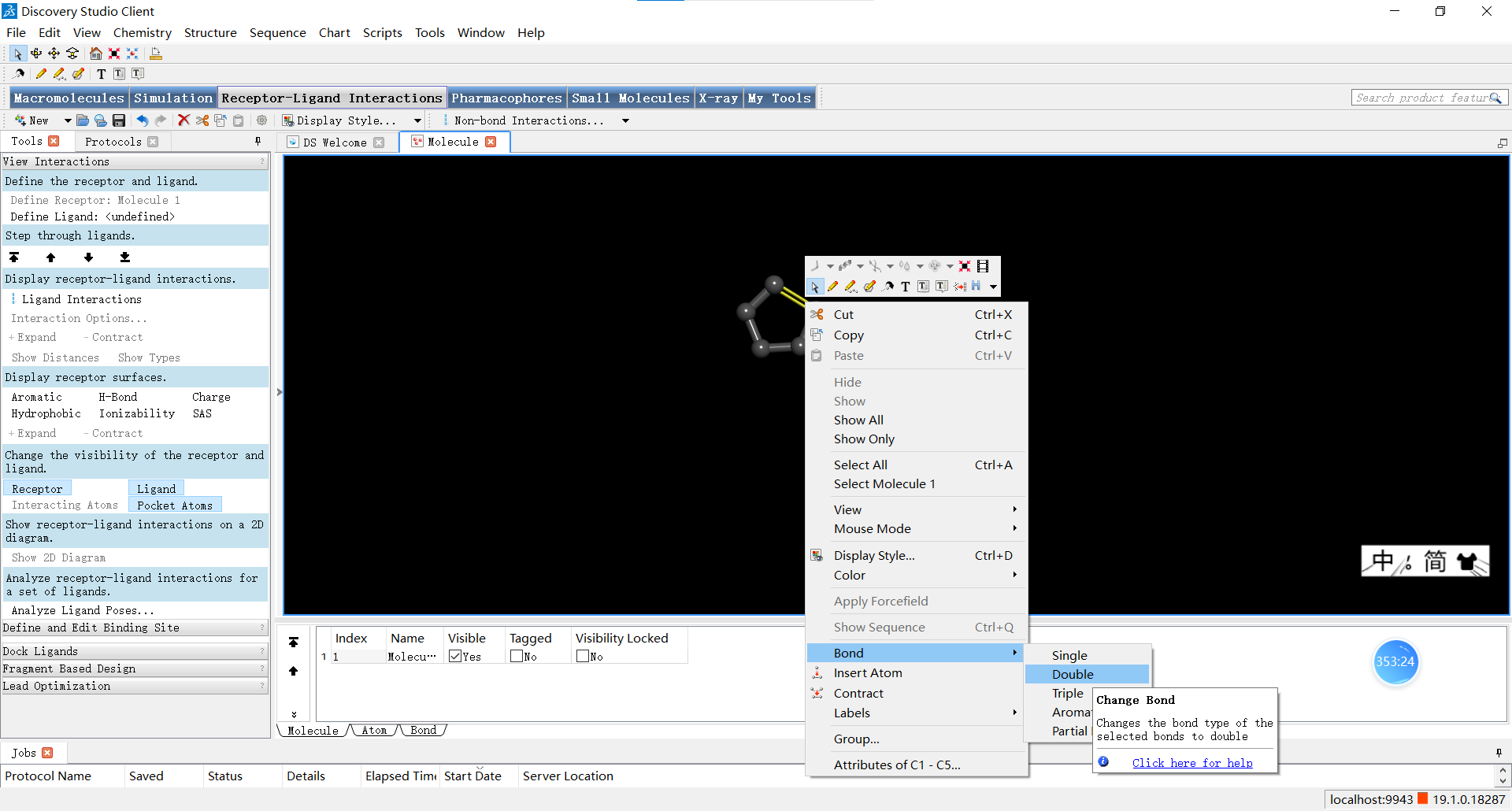
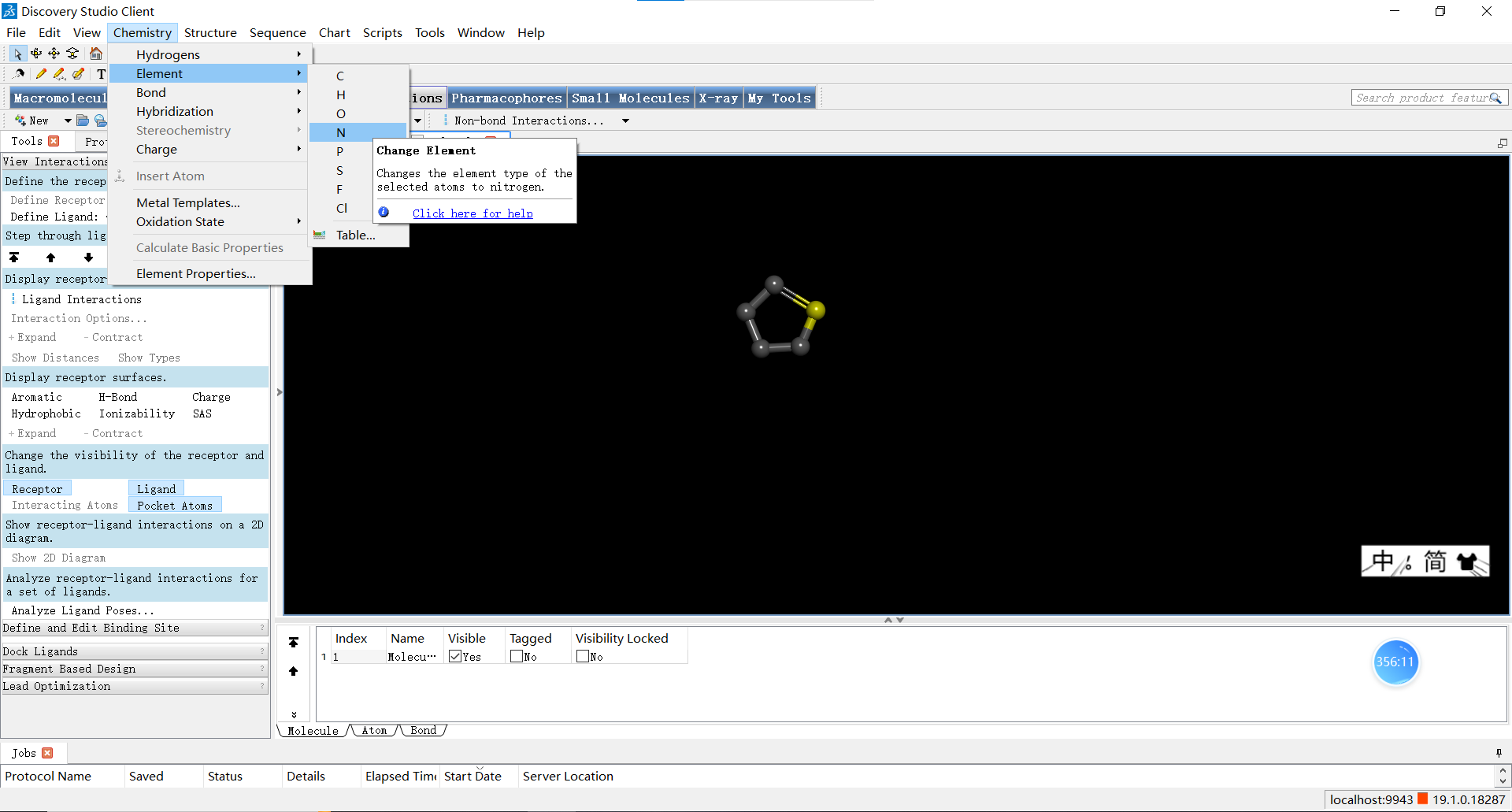
我们也可以选中某个原子或化学键后,点击上方的chemistry按钮拉开菜单,在这里可以进行添加氢原子、改变选中原子的元素类型、改变杂化轨道类型、改变原子化合价、改变原子氧化态等等操作,小伙伴们可以一一探索哦~小果在这里把选中的C原子更改成了N原子。
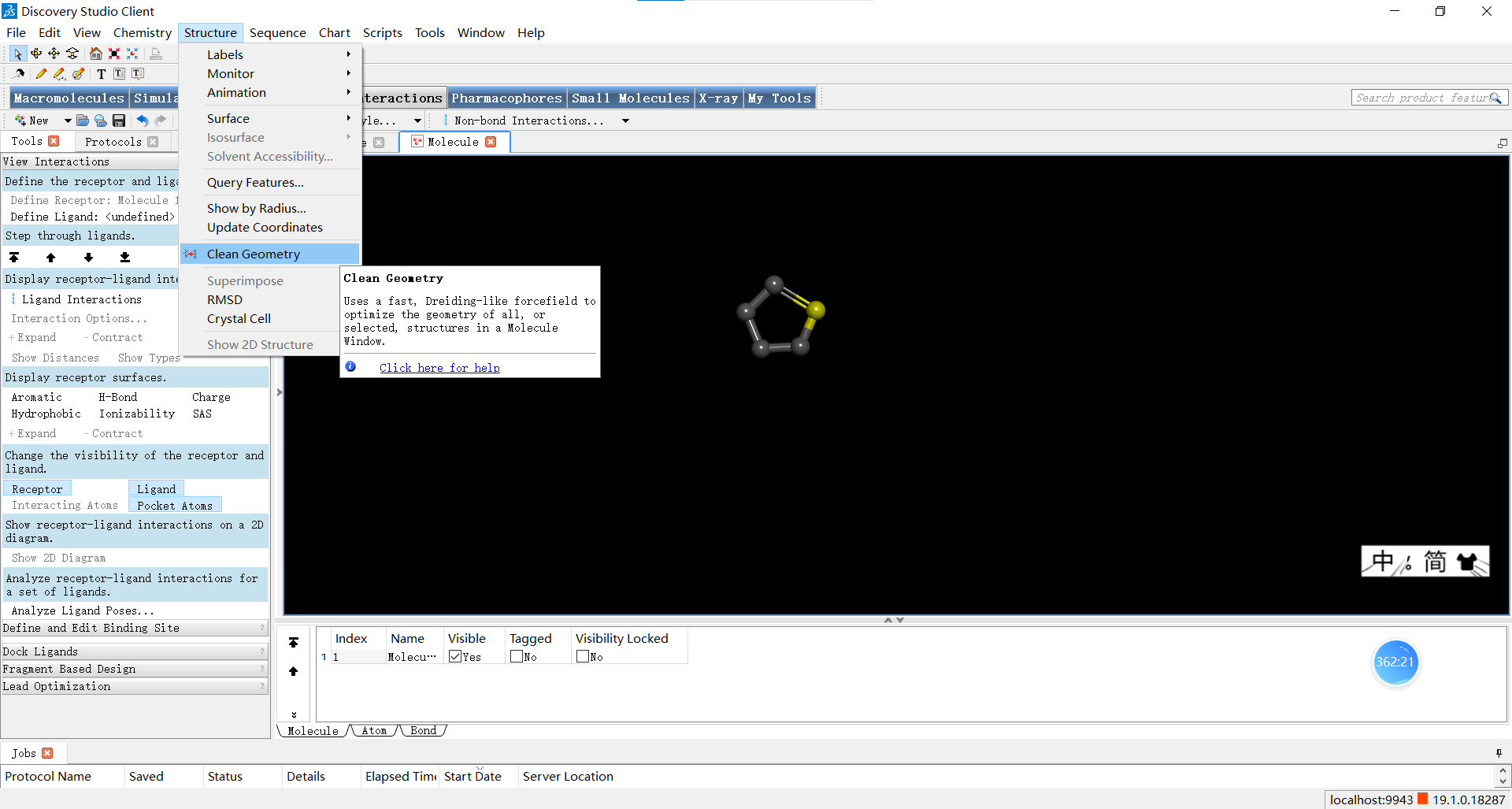
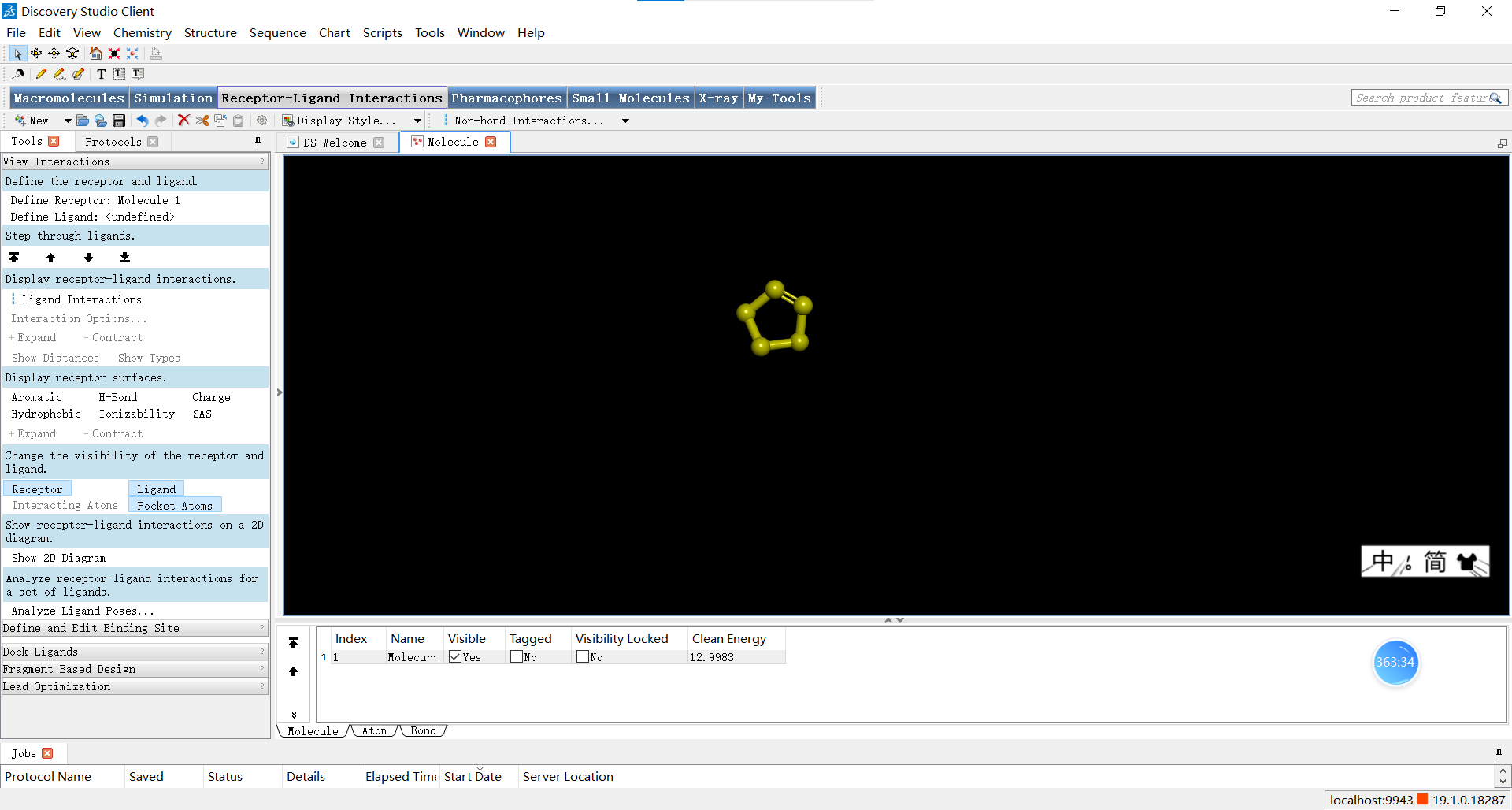
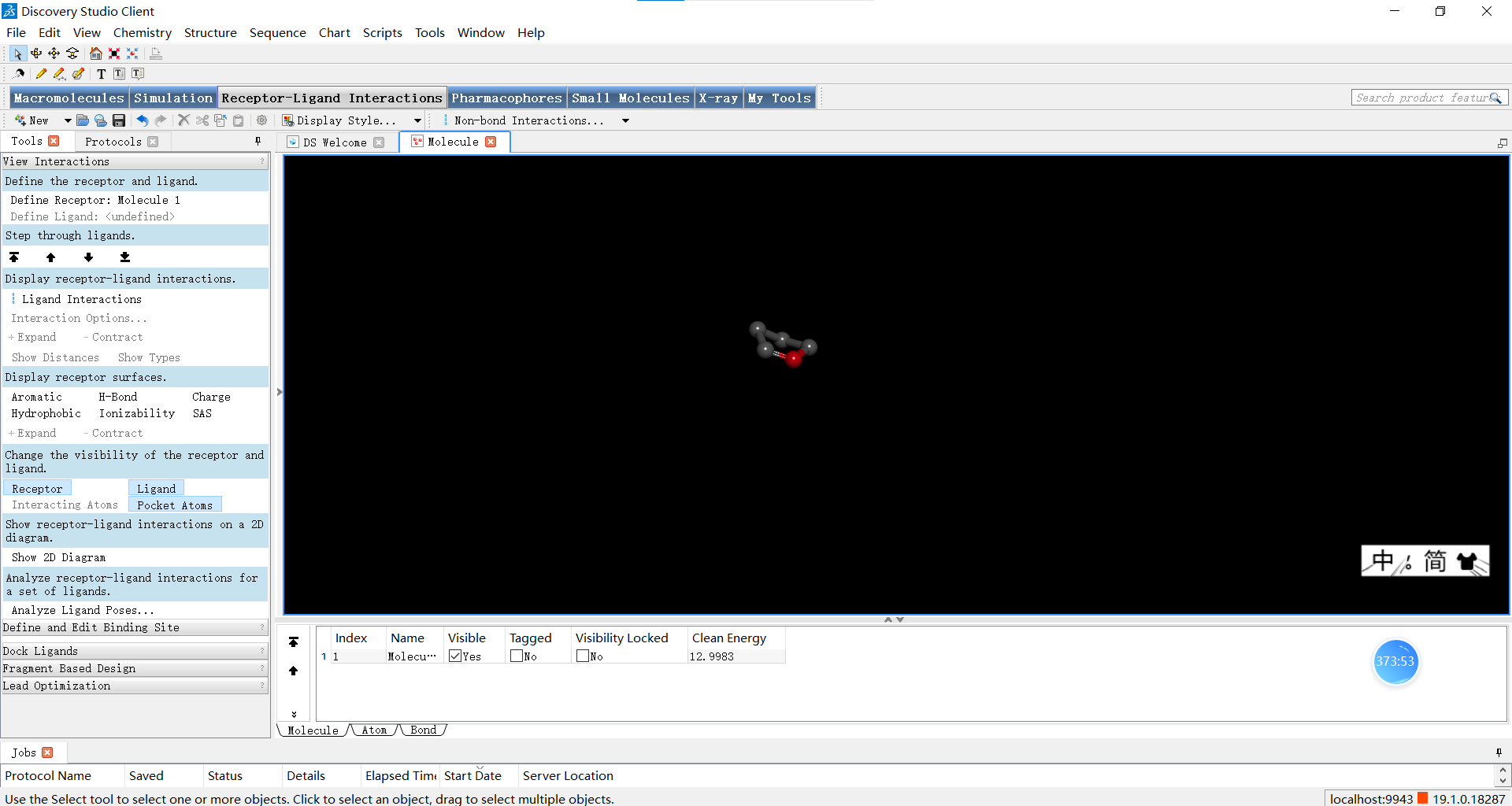
在小伙伴们调整完自己创建的小分子后,可以如图进行优化三维结构的操作,下面三张图分别是优化前,优化了选中的N原子后以及对整体分子进行优化后的结果,小伙伴们可以感受一下优化前后是不是确实有所不同呢~
我们也可以按住鼠标右键进行拖动来更换观察角度:
以上就是我们在Discovery studio自己绘制小分子配体并进行调整优化的过程啦,但是很多小伙伴可能会比较熟悉在别的平台或软件进行绘制小分子,或者直接导入现有的小分子来进行操作,那么下面小果就给大家展示一下如何对导入的比较复杂的小分子进行操作吧~
优化导入的配体
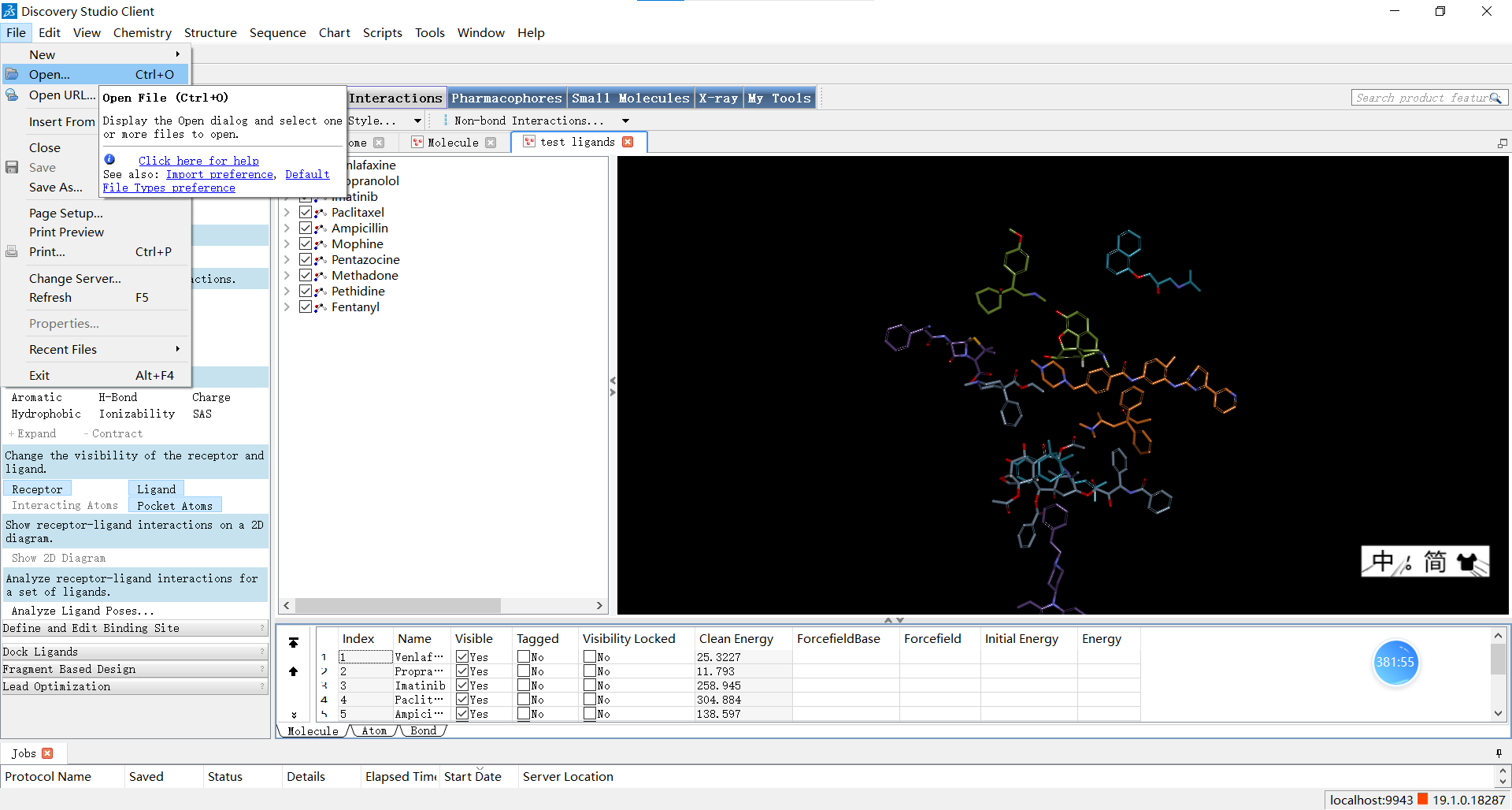
如图从file->open可以选择打开本地小分子文件
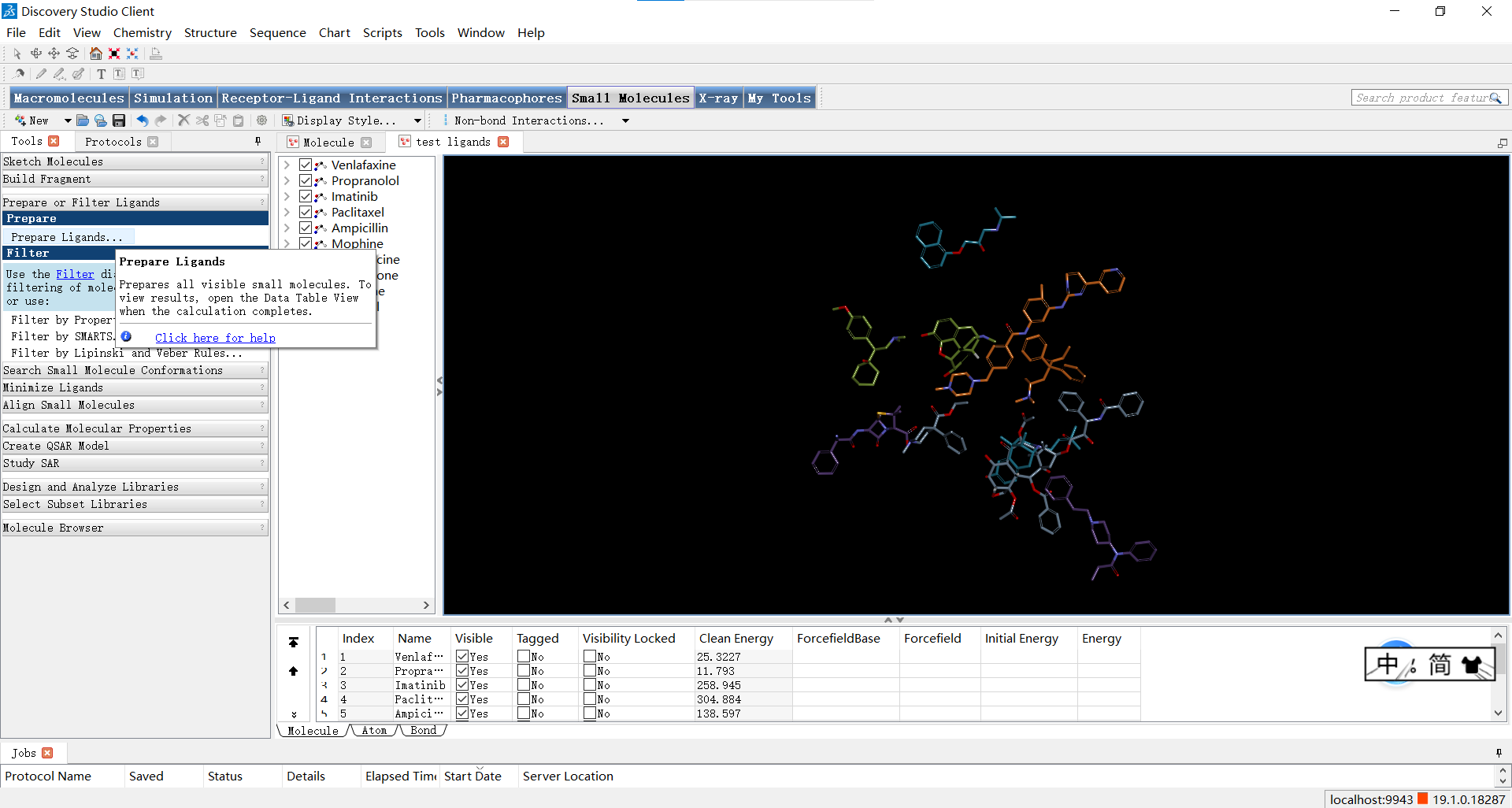
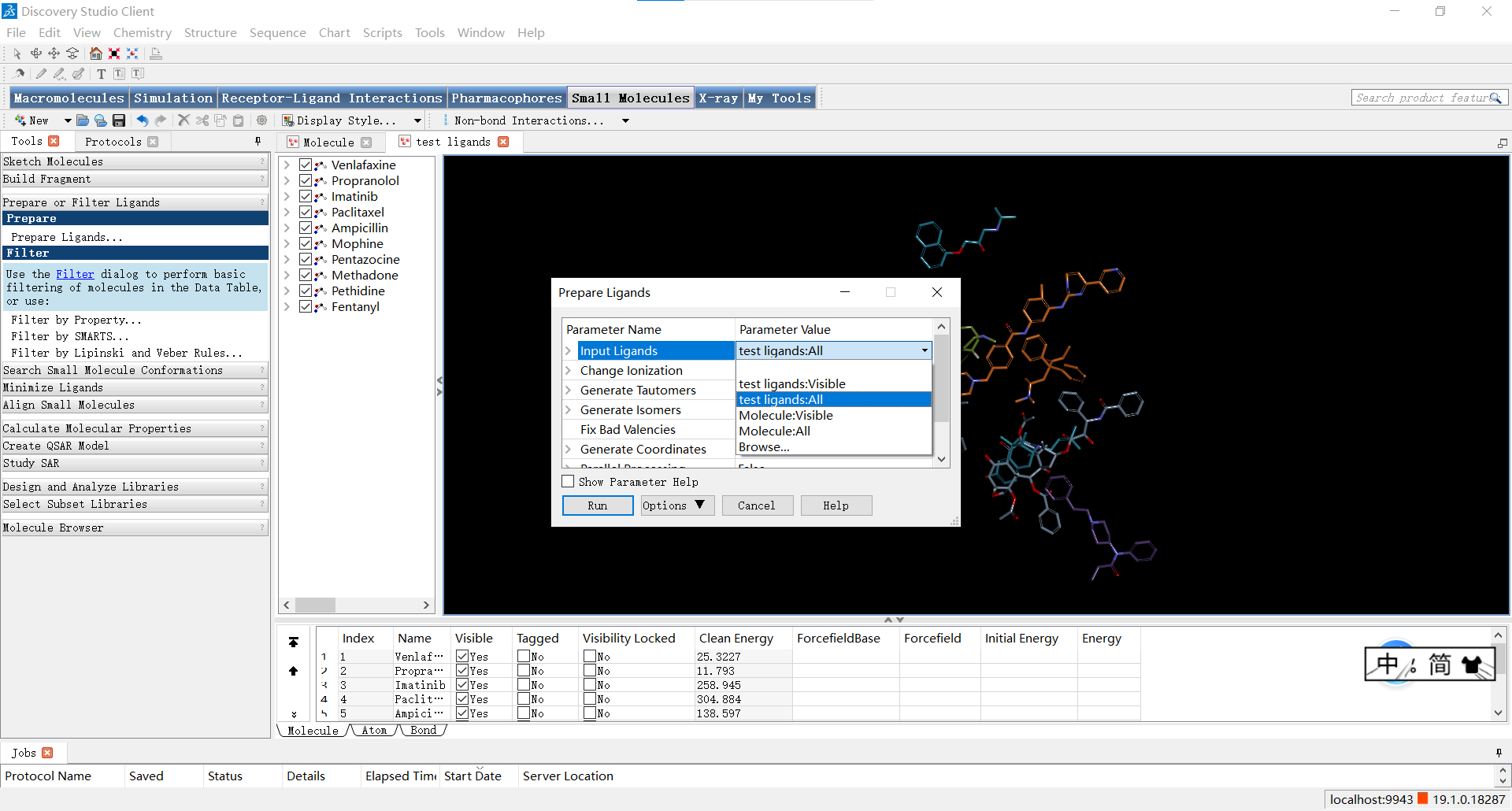
如图点击Small Molecules>Prepare or Filter Ligands>Prepare Ligands,可以打开参数设置面板;设置Input Ligands 参数为’文件名:All’,即表示对所有的小分子进行处理,其余参数一般默认,随后点击RUN运行。
运行结束后,我们可以在弹出的窗口点击report来查看处理信息,点击右侧的view result后就会弹出左侧的界面观察处理结果。
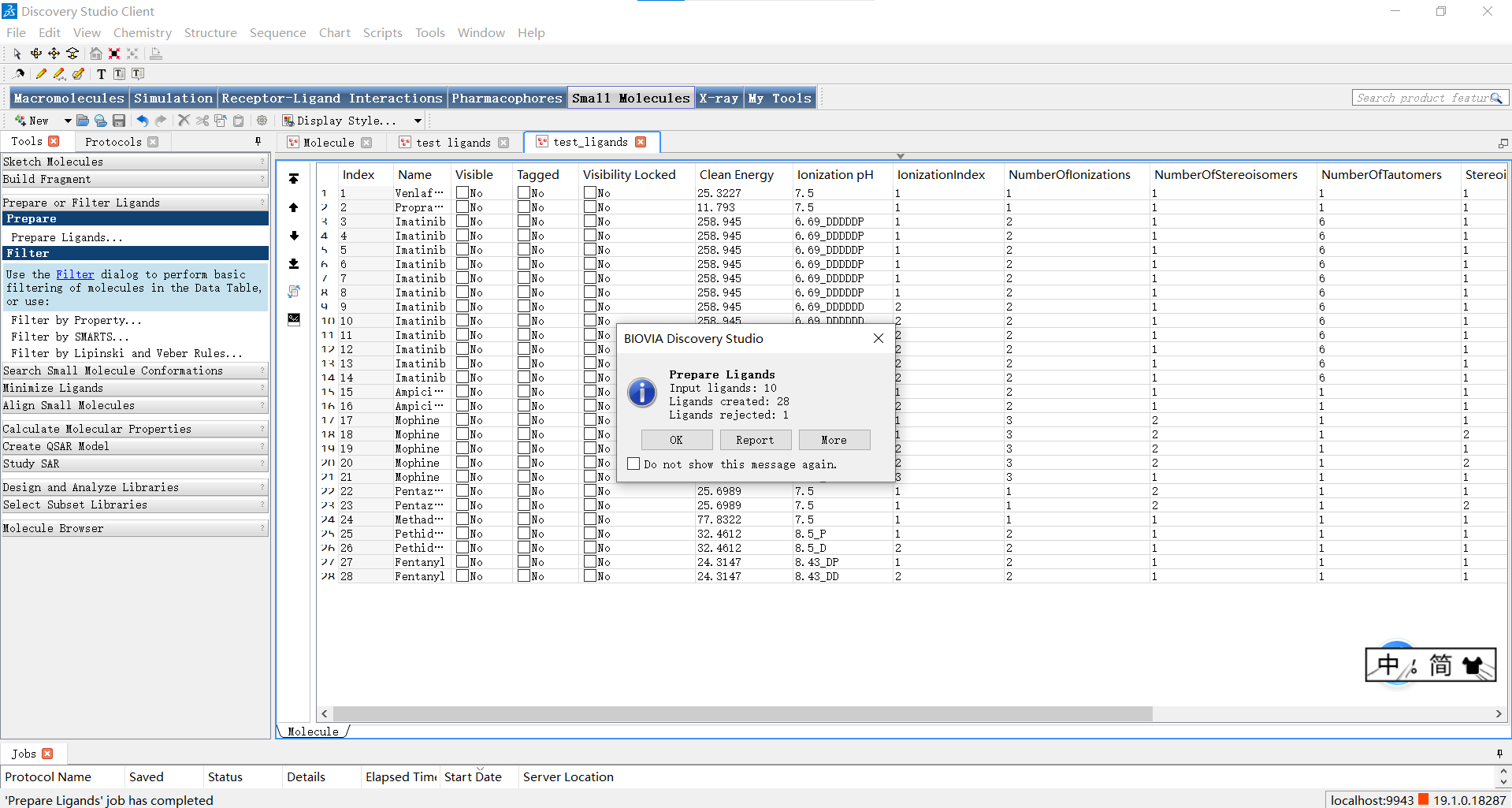
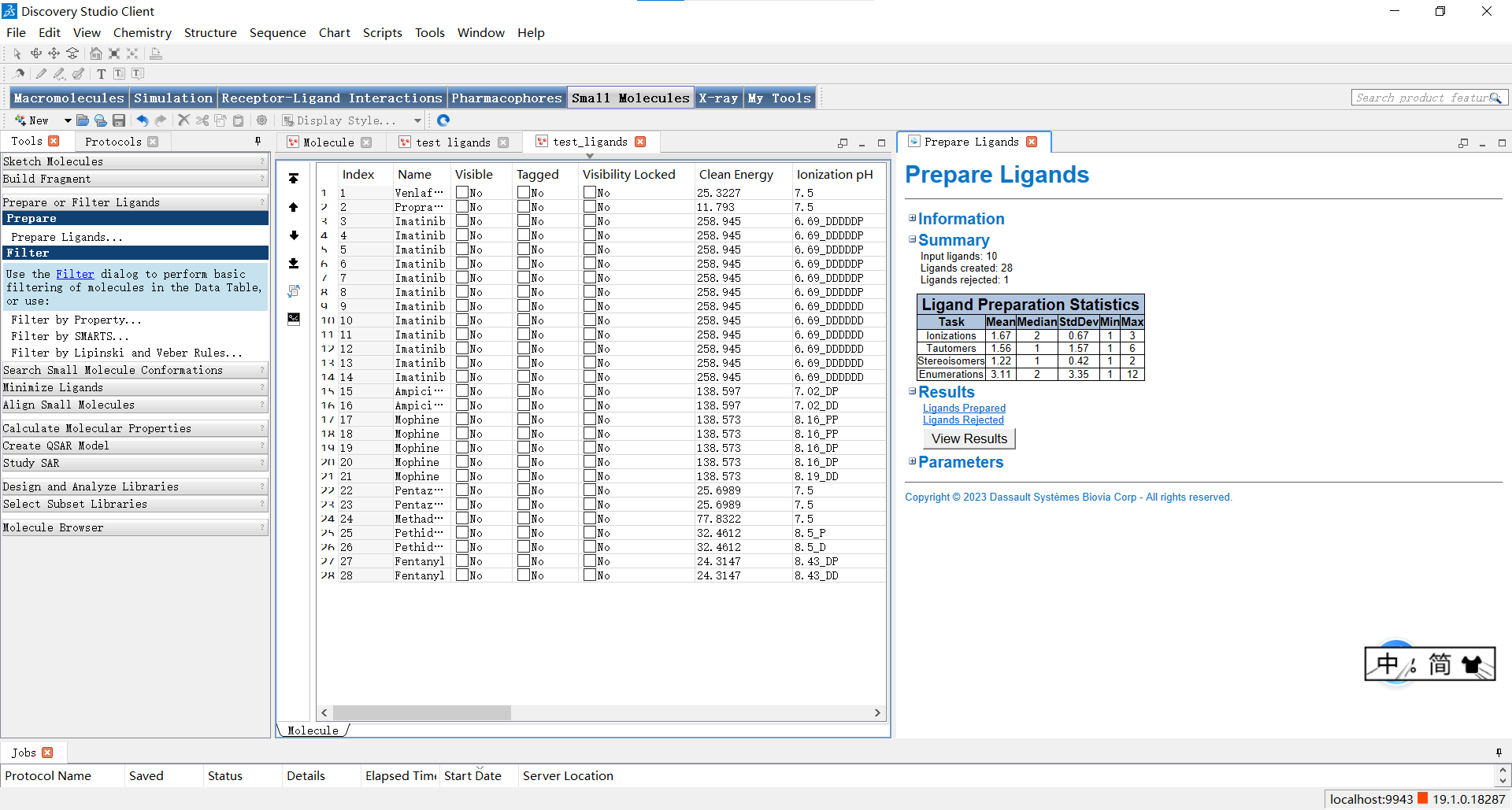
小伙伴们可能还不知道这一步操作有什么作用,小果在这里给大家解释一下:通过”Prepare Ligands”功能,可以对小分子进行以下操作:
- 清洁化合物结构:修复小分子中的结构问题,如修复键连接错误、填补缺失原子或修正原子的形式电荷等,确保分子结构的正确性和一致性。
- 添加氢原子:为了进行分子力场计算和其他相应计算,通常需要在小分子上添加适当的氢原子。”Prepare Ligands”功能可以自动添加氢原子,并调整其位置,以使得分子能够在力场计算中得到正确的描述。
- 离子化处理:该功能可以将小分子的离子状态进行修正,在模拟和计算过程中考虑溶剂和离子的效应。例如,可以对小分子进行质子化或去质子化处理。
- 能量最小化:”Prepare Ligands”还提供了能量最小化的选项,以进一步优化小分子的构象和性能。通过能量最小化,可以寻找小分子的最稳定构象,评估其能量和几何参数,并为后续计算提供更可靠的起点。
我们还可以通过Small Molecules|Minimize Ligands>Full Minimization按钮执行小分子的全能量最小化计算,调整原子的位置和键长、角度等参数,帮助优化分子的几何形状和空间排布,寻找小分子的最稳定构象。
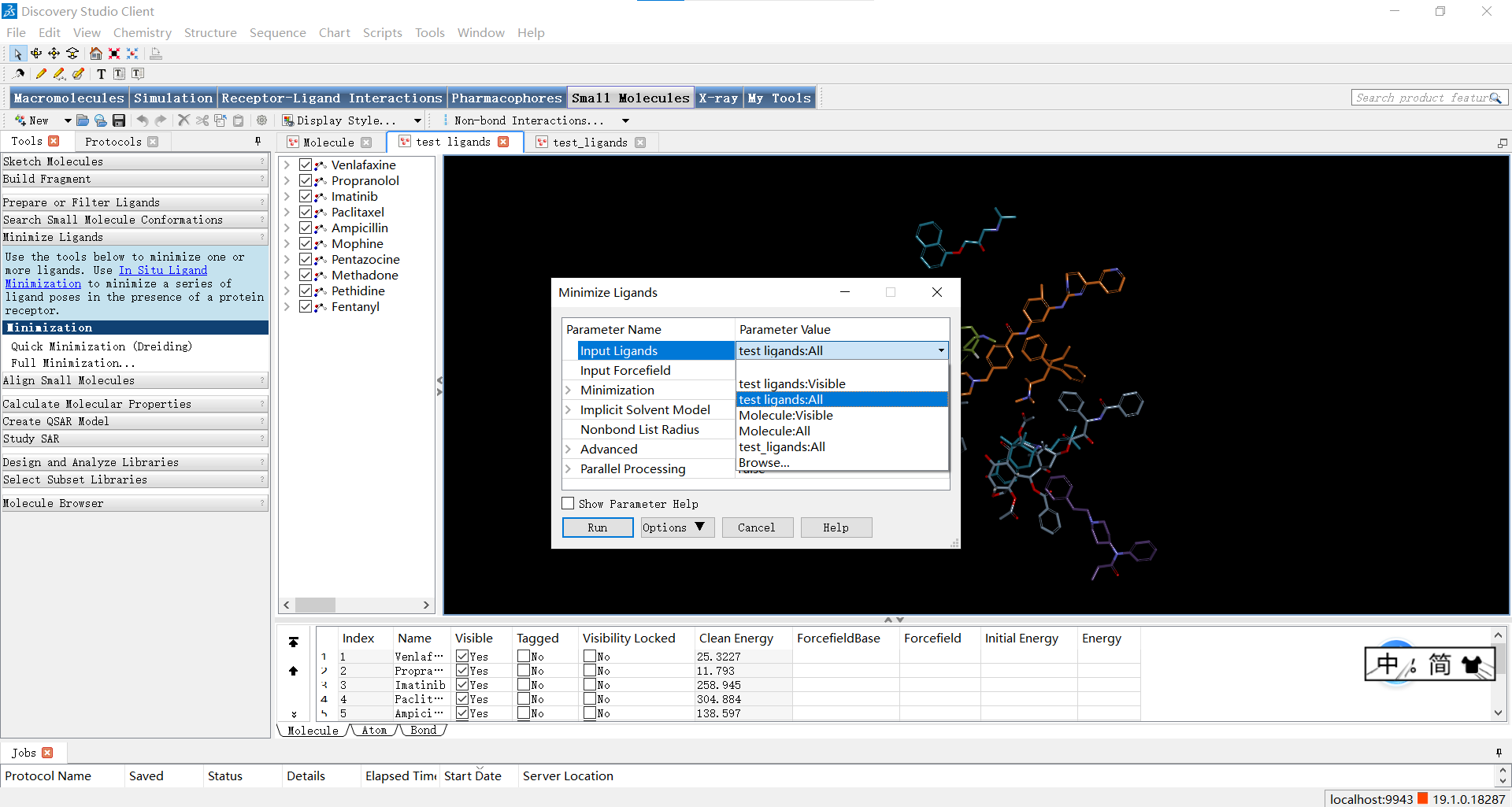
依然是设置Input Ligands 参数为’文件名:All’,其余参数默认,随后点击RUN运行。
运行结束后,我们可以从软件最下方的jobs中找到我们进行过的操作,点击即可打开已经完成过的操作结果啦。
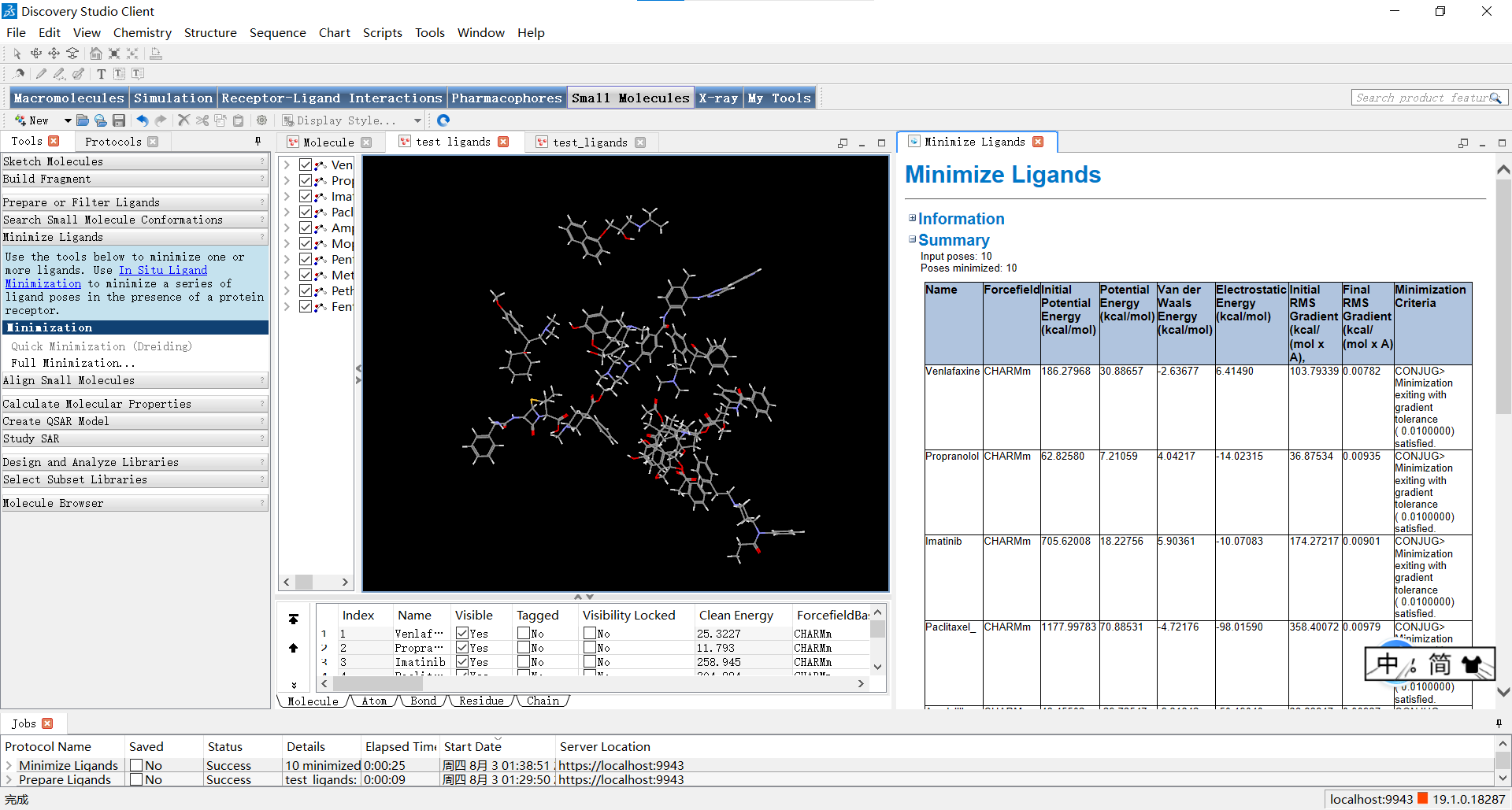
小伙伴们可以点击右侧窗口最下面的view results来更直观地观看结果,随后就会弹出左侧结果窗口,我们也可以进行拖动等操作来观察结果。
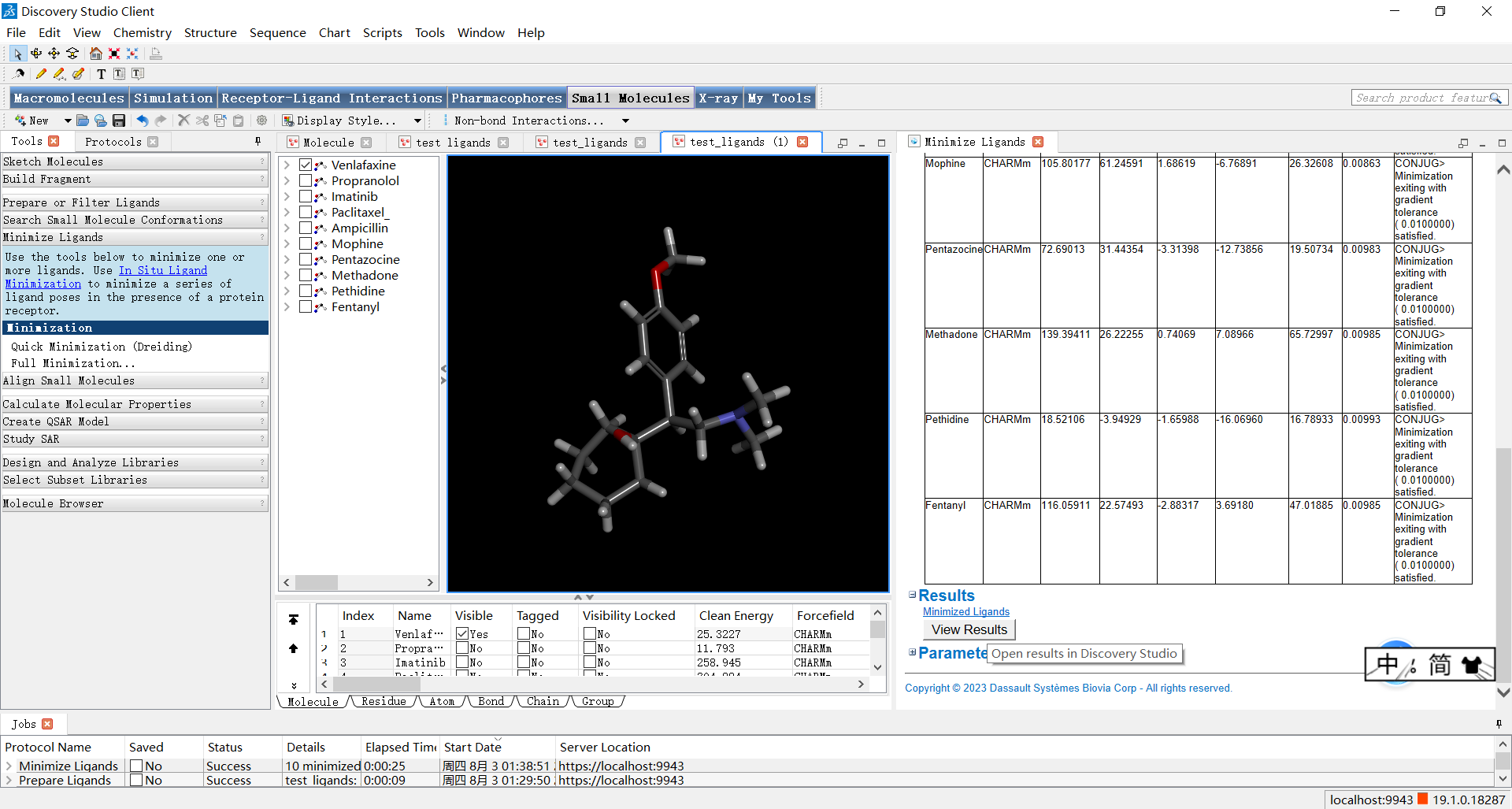
到这里为止,配体设计的最主要流程就已经完成啦,怎么样,是不是非常简单呢?细心的小伙伴可能已经注意到,当我们把鼠标放到discovery studio的大多数按钮上时,会出现help按钮,在使用过程中小伙伴们如果有什么疑问一定要多查看help哦,只有深入了解原理和具体操作步骤才能完成合格的药物设计。
本次的分享就到这里啦,如果小伙伴们平时在生信分析的操作过程中遇到困难,欢迎大家使用小果开发的生信工具平台http://www.biocloudservice.com/home.html哦。下一次的分享中小果会向大家介绍如何对蛋白结构进行处理,小伙伴们千万不要错过哦,小果期待与大家下次再见,拜拜~┏(^0^)┛