小伙伴在做生信分析时候,经常使用的流程就是筛选基因了,而到了最后筛基因的步骤却不知道如何去验证基因,往往使用的是预后的分析,却不了解某个基因的功能以及调控的通路。而一般在前面做差异基因时候就做了通路分析和GO功能富集,也只是整体基因的调控和功能。这个时候小伙伴想做单个基因的分析,就提问单基因的通路富集怎么做,小果在这里教小伙伴如何去做单基因的GSEA,让小伙伴了解自己筛选基因在那些调控通路以及其分子功能。
不过在这里小果提醒小伙伴一下,我们并不是对单个基因做GSEA,是拿我们单个基因相关的基因富集作为分组的方式,去探究我们想要分析基因的相关调控的通路。
跟着小果去学习吧
library(ggplot2)
#BiocManager::install(“limma”)
library(limma)
library(pheatmap)
library(ggsci)
lapply(c(‘clusterProfiler’,’enrichplot’,’patchwork’), function(x) {library(x, character.only = T)})
library(org.Hs.eg.db)
library(patchwork)
在这里需要以上几个包,小伙伴没下载的需要下载一下,其中org.Hs.eg.db就是做富集的包。但是可能不太好下载,用这个代码BiocManager::install试试。
小果这里用自己示例数据,小伙伴根据情况自行设置:
接下来
#输入文件
expFile=”after_merge.txt”
sgene=”GINS2″ #这里输入进行单基因GSEA的基因名称
下面我们对表达矩阵处理一下,先读入
rt=read.table(expFile,sep=”\t”,header=T,check.names=F)
rt=as.matrix(rt)
rownames(rt)=rt[,1]#把矩阵第一列放在R中的第一列
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)
接下来我们按照基因的表达分组
#按基因表达分组
group <- ifelse(data[c(sgene),]>median(data[c(sgene),]), “High”, “Low”)
group <- factor(group,levels = c(“High”,”Low”))
我们这里是是没有做差异分析的矩阵,我们需要找到与我们想要基因相关的,并且我们的基因也需要有差异才可以真实性
接下来:
#差异分析
design <- model.matrix(~0+group)
colnames(design) <- levels(group)
fit <- lmFit(data,design)
cont.matrix<-makeContrasts(High-Low,levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
deg=topTable(fit2,adjust=’fdr’,number=nrow(data))
Diff=deg
#保存单基因分组的所有基因差异结果
DIFFOUT=rbind(id=colnames(Diff),Diff)
write.table(DIFFOUT,file=paste0(“1.”,”DIFF_all.xls”),sep=”\t”,quote=F,col.names=F)
#展示差异最大的前30个基因
Diff=Diff[order(as.numeric(as.vector(Diff$logFC))),]
diffGene=as.vector(rownames(Diff))
diffLength=length(diffGene)
afGene=c()
if(diffLength>(60)){
afGene=diffGene[c(1:30,(diffLength-30+1):diffLength)]
}else{
afGene=diffGene
}
afExp=data[afGene,]
我们对分组进行标记,
#分组标签
Type1=as.data.frame(group)
Type1=Type1[order(Type1$group,decreasing = T),,drop=F]
Type=Type1[,1]
names(Type)=rownames(Type1)
Type=as.data.frame(Type)
#分组标签的注释颜色
anncolor=list(Type=c(High=”red”,Low=”blue” ))
pdf(file=paste0(“1.”, “DIFF_heatmap.pdf”),height=7,width=6)
pheatmap(afExp[,rownames(Type1)], #热图数据
annotation=Type, #分组
color = colorRampPalette(c(“blue”,”white”,”red”))(50), #热图颜色
cluster_cols =F, #不添加列聚类树
show_colnames = F, #展示列名
scale=”row”,
fontsize = 10,
fontsize_row=6,
fontsize_col=8,
annotation_colors=anncolor
)
dev.off()
#火山图差异标准设置,这个小伙伴自行设置,并不是一定要绘制
adjP=0.05
aflogFC=0.5
Significant=ifelse((Diff$P.Value<adjP & abs(Diff$logFC)>aflogFC), ife
#开始绘制
p = ggplot(Diff, aes(logFC, -log10(P.Value)))+
geom_point(aes(col=Significant),size=4)+ #size点的大小
scale_color_manual(values=c(pal_npg()(2)[2], “#838B8B”, pal_npg()(1)))+
labs(title = ” “)+
theme(plot.title = element_text(size = 16, hjust = 0.5, face = “bold”))+
geom_hline(aes(yintercept=-log10(adjP)), colour=”gray”, linetype=”twodash”,size=1)+
geom_vline(aes(xintercept=aflogFC), colour=”gray”, linetype=”twodash”,size=1)+
geom_vline(aes(xintercept=-aflogFC), colour=”gray”, linetype=”twodash”,size=1)
#查看,不添加标记可以直接保存
p
#添加基因点标记,按照,可自行根据差异分析的结果进行标记
point.Pvalue=0.01
point.logFc=1.5
其实以上小果也教了小伙伴火山图的绘制
我们直接读取结果,用下面结果去分析
logFC_t=0
deg$g=ifelse(deg$P.Value>0.05,’stable’,
ifelse( deg$logFC > logFC_t,’UP’,
ifelse( deg$logFC < -logFC_t,’DOWN’,’stable’) )
)
table(deg$g)
deg$symbol=rownames(deg)
df <- bitr(unique(deg$symbol), fromType = “SYMBOL”,
toType = c( “ENTREZID”),
OrgDb = org.Hs.eg.db)
DEG=deg
DEG=merge(DEG,df,by.y=’SYMBOL’,by.x=’symbol’)
data_all_sort <- DEG %>%
arrange(desc(logFC))
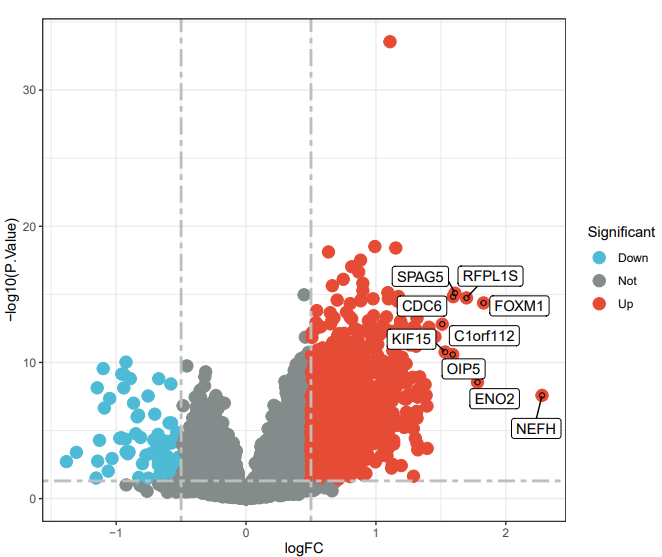
上述就是将差异的基因绘制成火山图,而我们单独做的基因也包括在内
geneList = data_all_sort$logFC #把foldchange按照从大到小提取出来
names(geneList) <- data_all_sort$ENTREZID #给上面提取的foldchange加上对应上ENTREZID
head(geneList)
上述就是差异分析的过程,接下来就是我们的重头戏:
#开始富集分析
kk2 <- gseKEGG(geneList = geneList,
organism = ‘hsa’,
nPerm = 10000,
minGSSize = 10,
maxGSSize = 200,
pvalueCutoff = 0.05,
pAdjustMethod = “none” )
class(kk2)
colnames(kk2@result)
kegg_result <- as.data.frame(kk2)
rownames(kk2@result)[head(order(kk2@result$enrichmentScore))]
af=as.data.frame(kk2@result)
write.table(af,file=paste0(“2.”,”all_GSEA.xls”),sep=”\t”,quote=F,col.names=T)#这里我们把结果保存一下
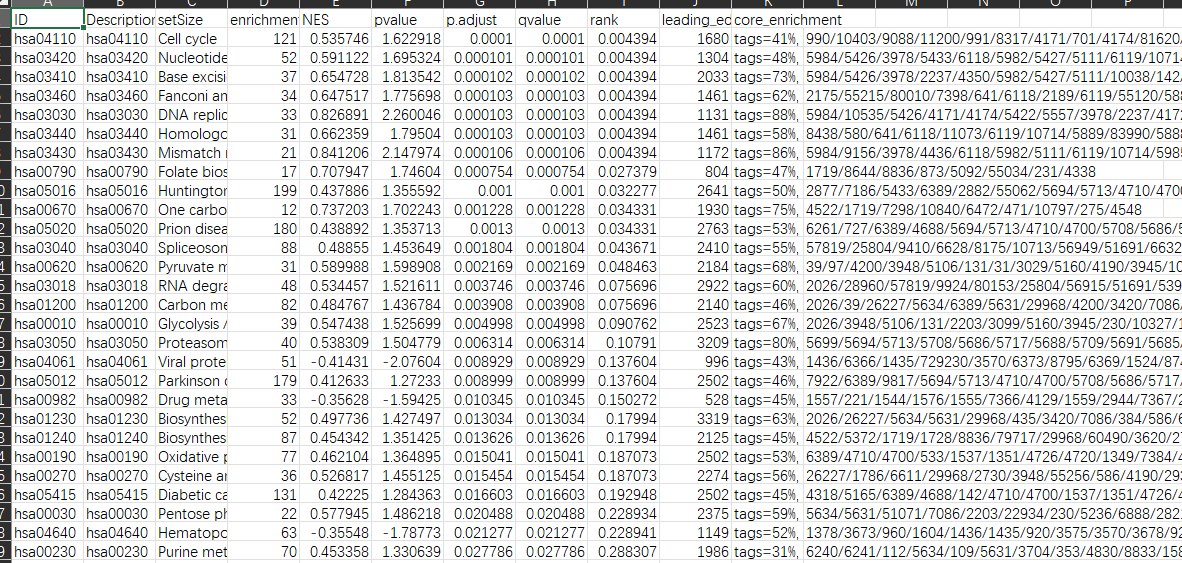
这个就是结果,下面我将他可视化!
#排序后分别取GSEA结果的前5个和后5个
num=5
pdf(paste0(“2.”,”down_GSEA.pdf”),width = 8,height = 8)
gseaplot2(kk2, geneSetID = rownames(kk2@result)[head(order(kk2@result$enrichmentScore),num)])
dev.off()
pdf(paste0(“2.”,”up_GSEA.pdf”),width = 8,height = 8)
gseaplot2(kk2, geneSetID = rownames(kk2@result)[tail(order(kk2@result$enrichmentScore),num)])
dev.off()
这里将上调的和下调的分析出图
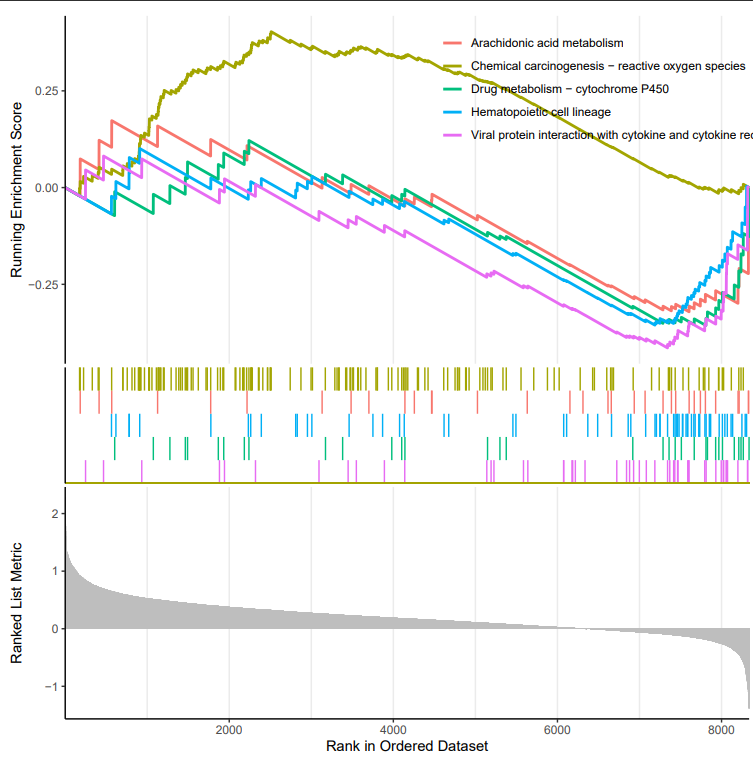
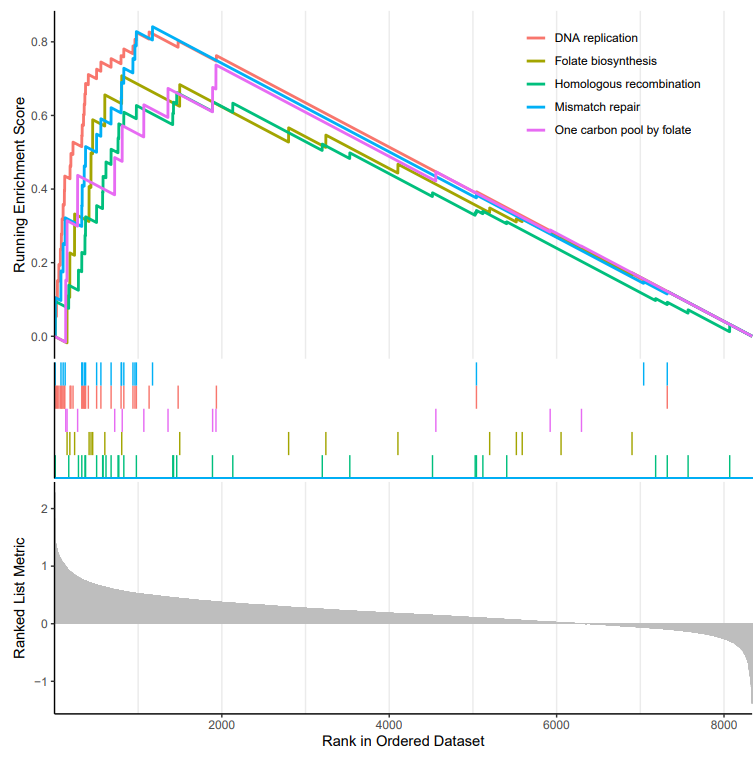 也可将所以的都放在一起
也可将所以的都放在一起
#排序后取前5个和后5个一起展示
num=5
pdf(paste0(“2.”,”all_GSEA.pdf”),width = 10,height = 10)
gseaplot2(kk2, geneSetID = rownames(kk2@result)[c(head(order(kk2@result$enrichmentScore),num),tail(order(kk2@result$enrichmentScore),num))])
dev.off()
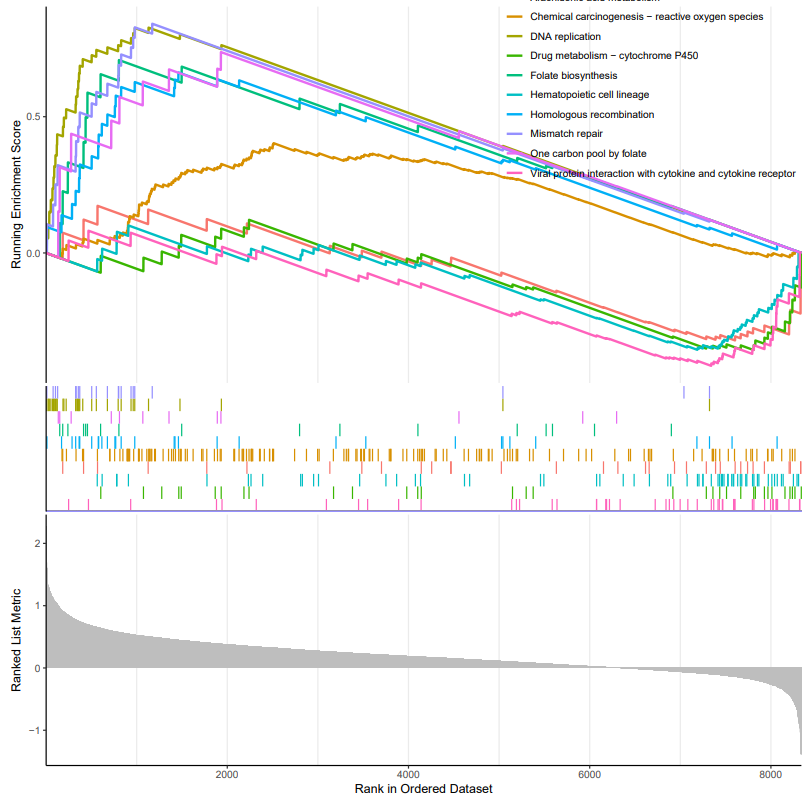
我们还可以其它的形式去展示结果:
这例我们将单独富集的#单独展示,小伙伴自行保存
gseaplot2(kk2,
title = “Th1 and Th2 cell differentiation”, #设置标题
“hsa04658″, #绘制hsa04658通路的结果,通路名称与编号对应
color=”red”, #线条颜色
base_size = 20, #基础字体的大小
subzplots = 1:3,
pvalue_table = T) # 显示p值
小伙伴根据自己想要的通路去绘制吧
以上就是GSEA的绘制了,小伙伴有没有学会呢,绘制的代码其实不复杂,中间参杂了差异分析,看起很多,其实精髓都在后面的GSEA分析代码中,小伙伴要多多理解代码的意思,这样才能绘制出自己想要的图片