小伙伴们大家好,在之前的分享中小果详细地给大家介绍了小分子配体的准备和蛋白结构的处理,那么这一次小果就给大家带来配体和蛋白的对接方法。废话不多说,我们直接进入正题~
什么是分子对接?
分子对接(Molecular docking)是一种计算方法,用于预测蛋白质和小分子(配体)之间的结合方式和相互作用强度。它是药物设计、生物活性研究和分子识别中重要的工具之一。
分子对接的目标是预测蛋白质和配体在空间上的最佳相互排列,以寻找稳定的结合构象和判断结合强度。通过模拟这种结合过程,可以预测药物候选物与靶蛋白的相互作用模式,为药物发现和设计提供指导。
分子对接的步骤?
以下是小果给大家总结的分子对接一般步骤:
- 预处理:准备蛋白质和配体的结构,包括去除水分子、离子等、修正构象问题;添加氢原子等操作。这一步小果在之前的文章中给大家特别详细地分享过如何使用Discovery Studio进行操作,没有看过的小伙伴们记得去看一看哦~
- 定义活性位点:确定蛋白质上可能与配体相互作用的区域,即活性位点(或称受体结合位点)。活性位点的选择通常基于蛋白质的结构信息和相关生物活性数据。
- 定义搜索空间:根据活性位点确定一个合理的搜索空间,限制配体的探索范围,以提高计算效率。
- 对接搜索:使用适当的分子对接算法,在搜索空间内进行多个配体构象的快速探索,并评估它们与蛋白质的结合能。
- 结果分析和评估:对对接结果进行分析,包括预测的最佳配体构象、结合位点和相互作用模式等。使用评分函数对不同构象进行评估,判断其亲和性和稳定性。
- 结果优化:根据对接结果,可以进行后续的结构优化,包括引入修饰基团、设计药物衍生物等,以进一步改善结合亲和性和选择性。
小果提醒大家,在进行分子对接时,需要注意以下几个方面:
- 活性位点准确性:活性位点定义的准确性对于分子对接的结果至关重要。需要依赖蛋白质结构信息和生物活性数据来确定活性位点。
- 活性位点不完全等于受体结合位点:活性位点和受体结合位点在大多数情况下是相互重叠的,指的是同一个区域或位置。然而,它们可能在以下情况下有所不同:
- 多个结合位点:某些蛋白质可能具有多个结合位点,其中只有一个是活性位点,其他位点可能与配体结合,但没有明显的生物活性。因此,活性位点是在多个结合位点中具有特定功能的子区域。
- 构象变化:在某些情况下,蛋白质可以通过构象变化来改变其结合位点的位置或形状。这可能导致活性位点和受体结合位点不完全重叠,因为在特定构象下才能呈现出活性位点。
- 配体选择性:某些蛋白质可能具有与不同类别的配体结合的能力,并且这些配体与受体结合的位置可能略有不同。在这种情况下,活性位点可以指的是针对特定类别配体的优先选择区域。
- 灵活性处理:蛋白质和配体的灵活性(指它们在结合过程中可能发生的构象变化或柔性运动)在结合过程中起着重要作用,可以采用柔性对接方法或分子动力学模拟等技术来考虑灵活性。
- (注:蛋白质的灵活性通常指的是蛋白质的构象变化能力。蛋白质可以通过内部构象变化(如侧链旋转、片段移动等)或整体构象变化(如蛋白质的收缩、延伸、折叠等)来适应与配体的结合,这种灵活性是蛋白质功能的重要基础,使得蛋白质能够与多种不同结构和大小的配体发生结合。
- 而配体的灵活性通常指的是配体的构象变化或柔性运动。配体可能是小分子药物、底物或其他与蛋白质相互作用的小分子。配体的灵活性意味着它可以通过构象调整来适应与蛋白质的结合位点的形状、电荷分布和其他特征。这种灵活性使得配体能够与蛋白质形成稳定的结合,并发挥其生物活性或功能。)
- 参数设置:选择适当的算法和参数非常重要。不同的算法和评分函数可能适用于不同类型的结合系统,需要根据具体情况进行调整和验证。
- 结果解释与验证:分子对接结果应当谨慎解释。在最高评分的结合模式下,仍然需要考虑实验验证和进一步优化。
- 实验验证:分子对接是一种计算方法,在结果的解释和验证方面需要与实验相结合。实验技术如结晶学、核磁共振等可以提供更准确的结合模式和亲和性信息。
相信大家已经清楚了分子对接的基本步骤,那么小果现在就向大家介绍如何使用Discovery Studio进行分子对接~
如何定义活性位点?
小果在之前的文章中给大家特别详细地分享过如何使用Discovery Studio对小分子配体和蛋白结构进行处理,因此我们直接从定义活性位点这一步开始,没有看过前面分享的小伙伴们记得去看一看哦~
首先第一步我们需要将经过预处理的蛋白定义为受体,具体位置如下图所示:Receptor-Ligand Interactions>Define and Edit Binding Site>Define Receptor
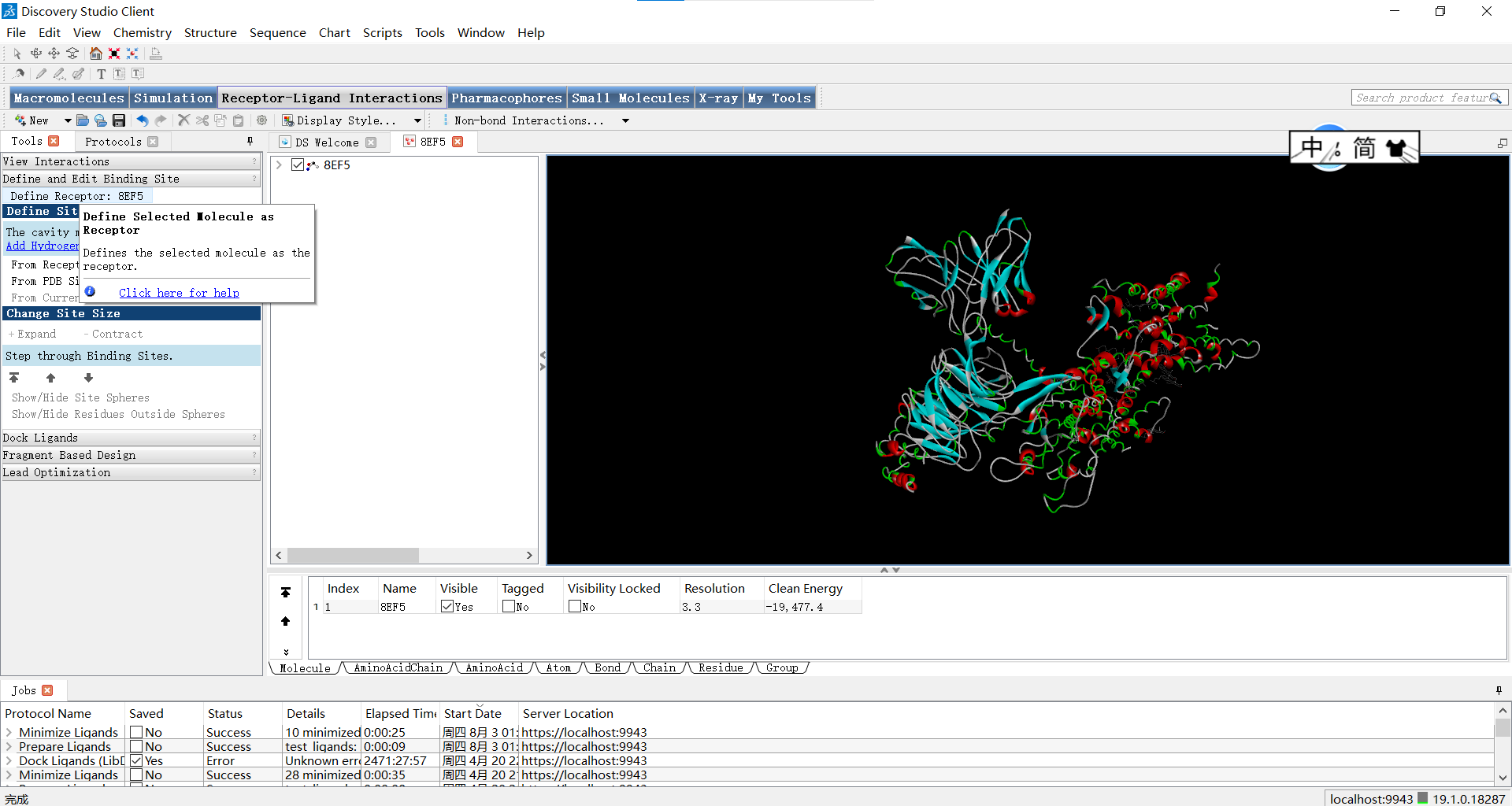
这一步操作完成之后,我们要选择定义哪里为活性位点。定义活性位点我们需要考虑以下几个因素:
- 保守性氨基酸:在蛋白质家族中,活性位点通常包含高度保守的氨基酸残基,这些残基在不同成员之间存在较低的变异率,这些氨基酸可能是关键的催化或配体结合残基。例如,血红蛋白家族中的活性位点通常包含高度保守的组氨酸残基(His)和亮氨酸残基(Leu)。
- 催化残基:活性位点常包含一些特定的催化残基,如亲核残基、酸或碱残基等,这些催化残基能够参与催化反应中的特定步骤,从而促进催化活性。例如丙酮酸激酶(Acetolactate synthase)的活性位点包含Lys残基,该残基在催化反应中起到碱催化的作用。
- 底物识别残基:活性位点上的某些氨基酸残基可能参与底物的识别和特异性结合,这些残基可以通过氢键、疏水相互作用、离子相互作用等方式与底物分子发生相互作用。例如乳酸脱氢酶(Lactate dehydrogenase)的活性位点上的Arg残基与底物丙酮酸之间形成氢键,从而识别并结合特定的底物。
- 金属离子配位位点:某些蛋白质需要金属离子来实现催化活性,活性位点中的特定氨基酸残基可以形成金属离子的配位位点,从而稳定金属离子并参与催化过程。例如类胰岛素样生长因子受体(Insulin-like growth factor receptor)的活性位点中通过His残基形成的配位位点可以稳定金属离子,进而参与催化过程。
- 氢键网络:活性位点中的氨基酸残基可以通过氢键形成特定的网络结构,这有助于保持位点的稳定性和功能活性。例如乙醇脱氢酶(Alcohol dehydrogenase)的活性位点中的氨基酸残基形成氢键网络,维持活性位点的稳定性和催化活性。
- 疏水口袋:某些活性位点可能包含疏水口袋,能够在催化过程中与疏水性底物或配体相互作用。例如乙酰胆碱酯酶(Acetylcholinesterase)的活性位点中的疏水口袋能够与底物乙酰胆碱分子的疏水部分相互作用。
- 活性位点的拓扑特征:活性位点的拓扑特征指的是空间结构和残基之间的关系。例如,凹槽、隧道或开放的面积等特征可以影响底物分子的进入和结合,蛋白酪氨酸激酶(Tyrosine kinase)的活性位点具有凹槽结构,该结构能够容纳和定位底物分子,从而实现催化反应。
这里可能有小伙伴反映说,参考这么多因素实在太麻烦了,我不需要做到这么完美,有没有简单点的方法?那么小果在这里给大家两个定义活性位点的建议:
首先,小伙伴们可以去查阅文献,关于这个蛋白是否有相应的研究来报道最适合定义为活性位点的区域在哪里。
如果没有相关的文献怎么办呢?小果再给大家推荐一个常用的方法,那就是通过Discovery Studio来分析哪里适合作为活性位点:
具体的操作按钮是Receptor-Ligand Interactions->Define and Edit Binding Site->From Receptor Cavities
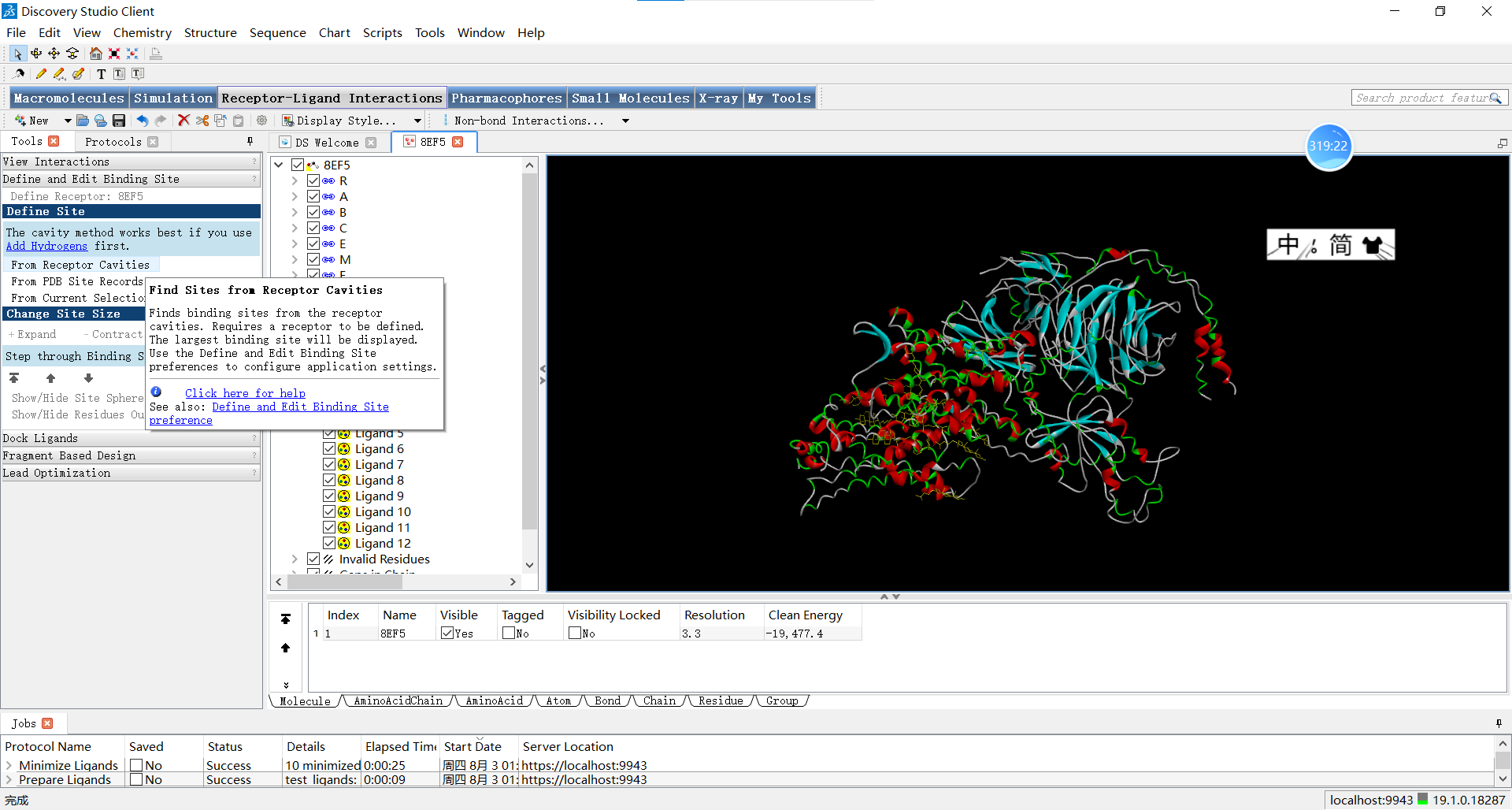
想要深入了解的小伙伴可以查询官方的文档:
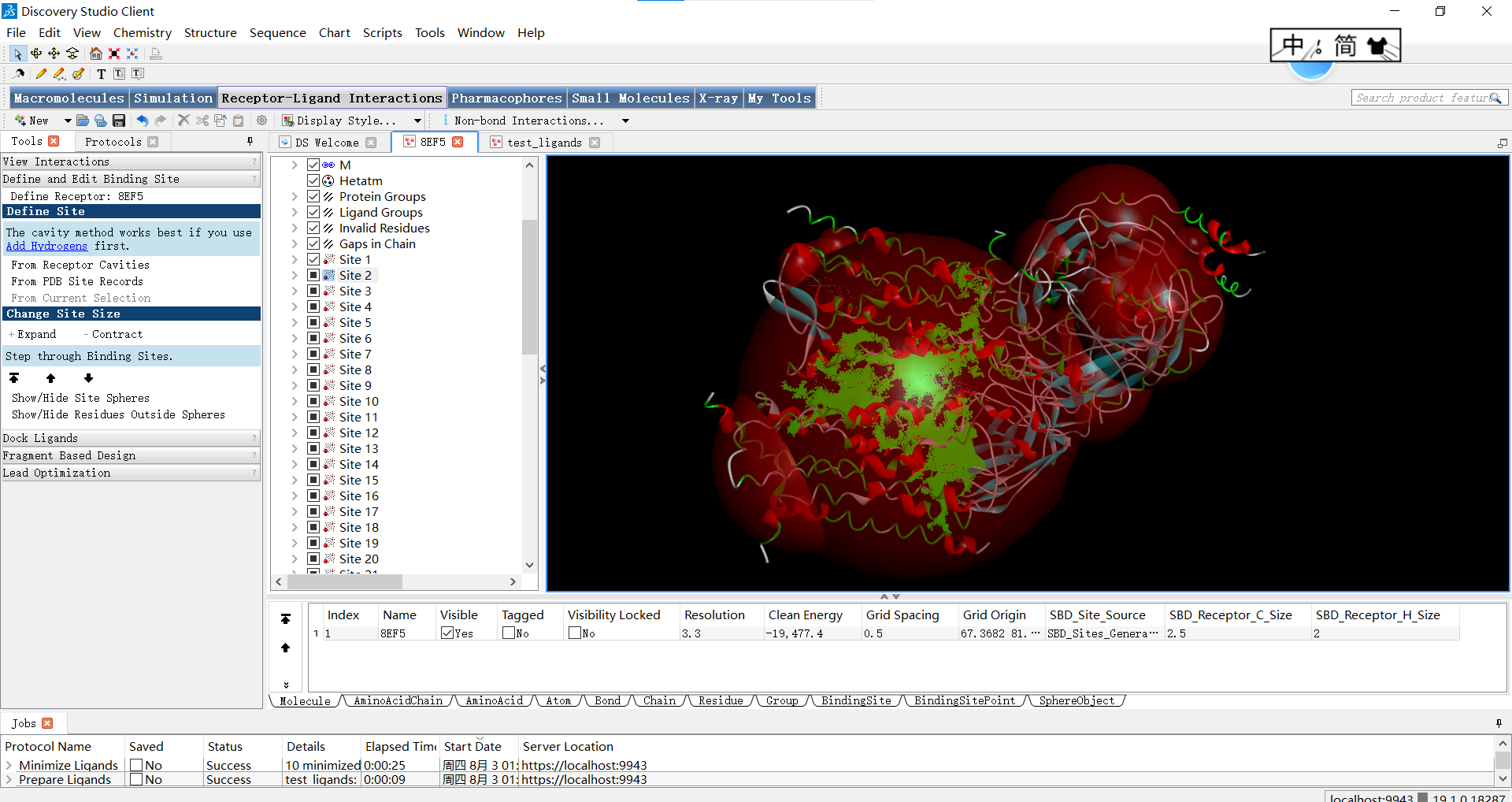
点击之后等待一段时间,软件会帮我们分析出推荐的位点,展示在左侧,我们可以挨个勾选来进行查看(注:红球内部即为活性位点):
全部位点:
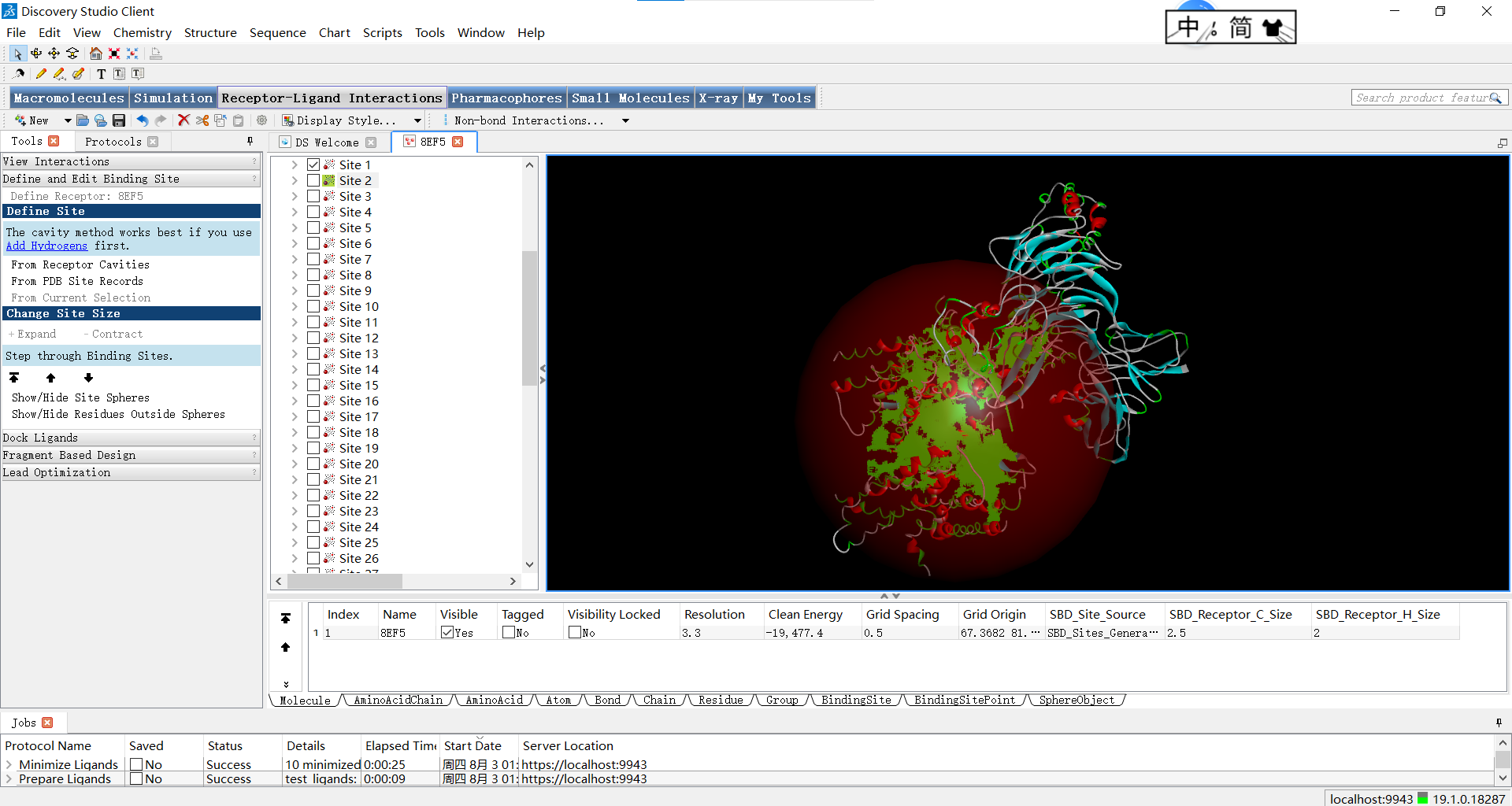
位点一:
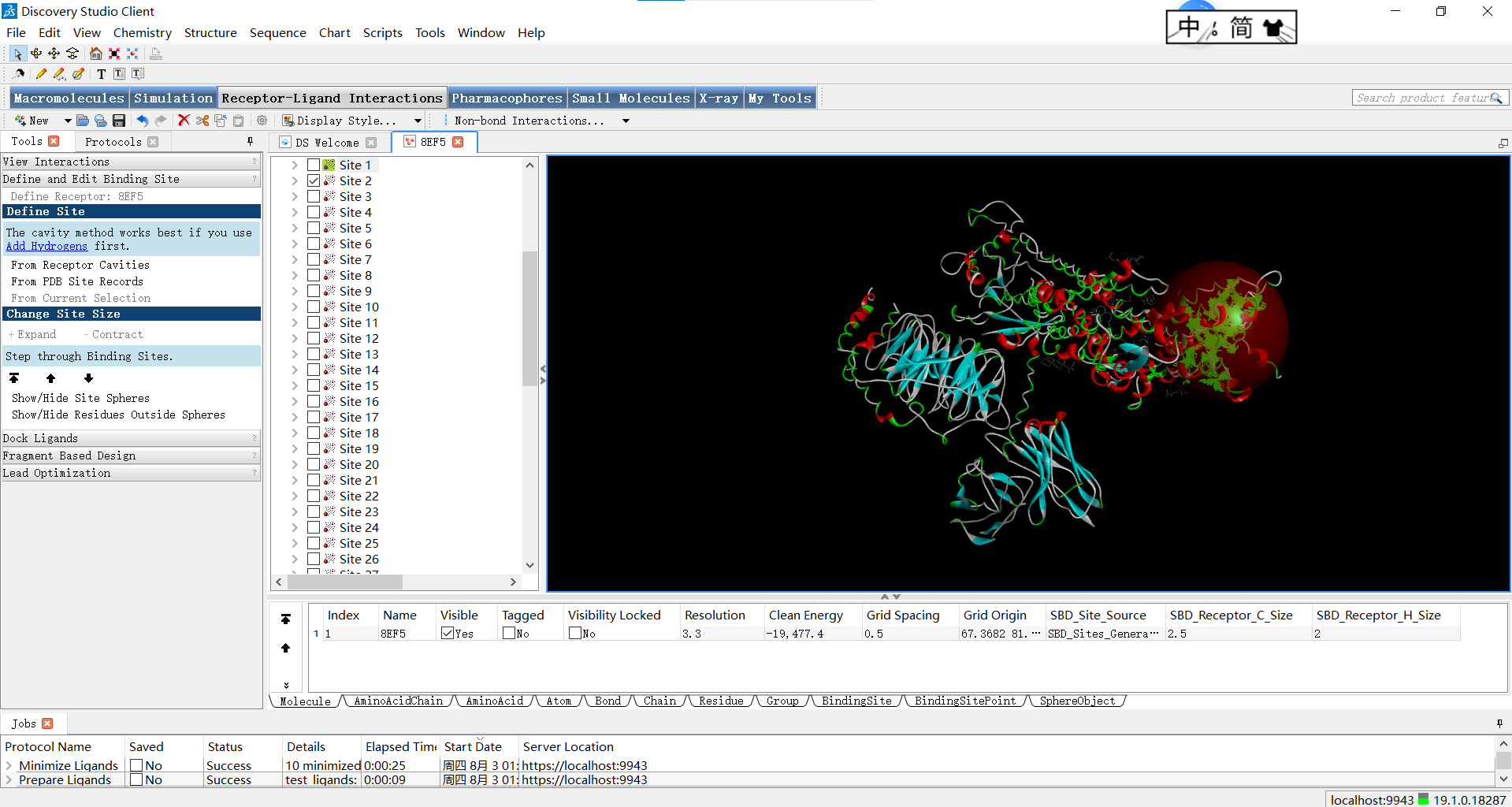
位点二:
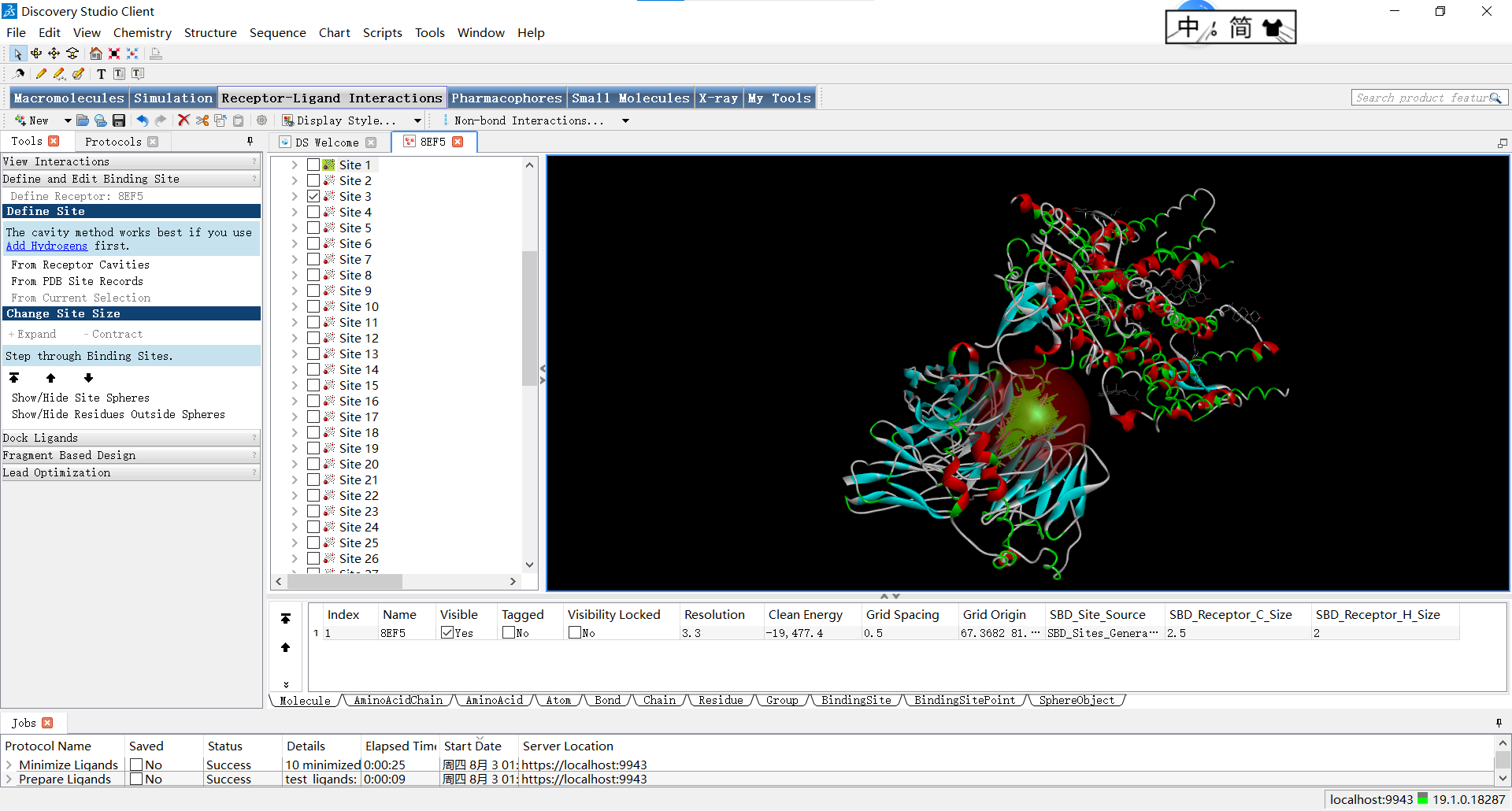
位点三:
如何查看活性位点?
上面的红球叫做SBD(Shape-Based Descriptors),它是SBD方法中的一种基于形状的描述符,用于表示分子的三维结构和表面特征,即描述蛋白质活性位点周围的球形体积。在蛋白质的活性位点中,通常存在着特定的空间要求和形状特征,以适配与其相互作用的小分子配体。SBD_Site_Sphere通过将位点周围的空间划分为多个球形体积来描述活性位点的形状特征。每个球形体积都可以具有不同的属性,例如半径、位置和物理化学特性。
通过使用SBD_Site_Sphere描述符,可以将蛋白质活性位点的形状信息转化为数值化的表示,以便进行计算和比较。这些描述符可用于药物设计、虚拟筛选和活性位点预测等应用。下面小果再教大家几招观察和调整SBD_Site_Sphere的方法:
Show/Hide Site Spheres 可以控制是否显示红球
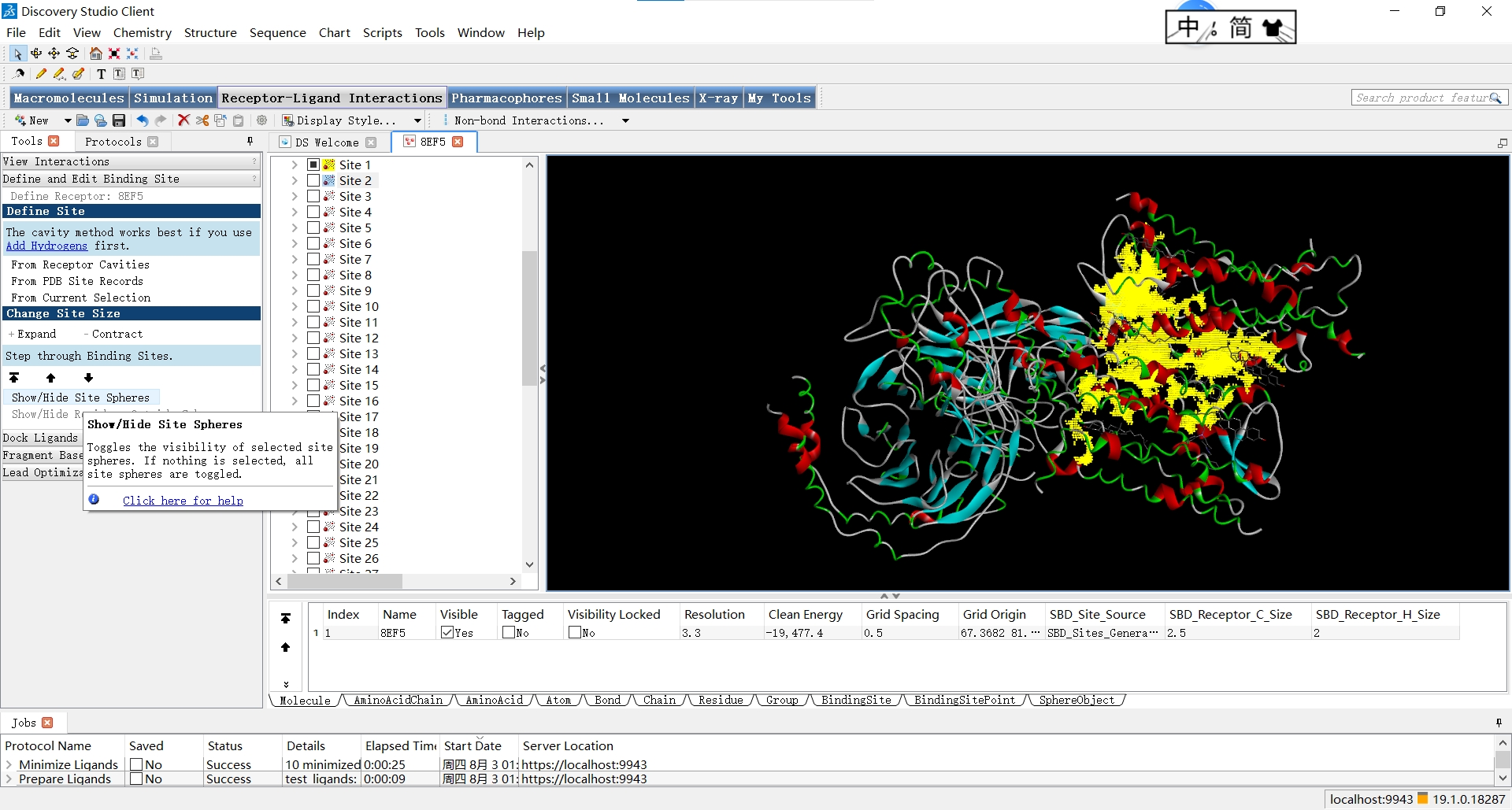
通过change site size下的contract和expand可以调整红球的大小:
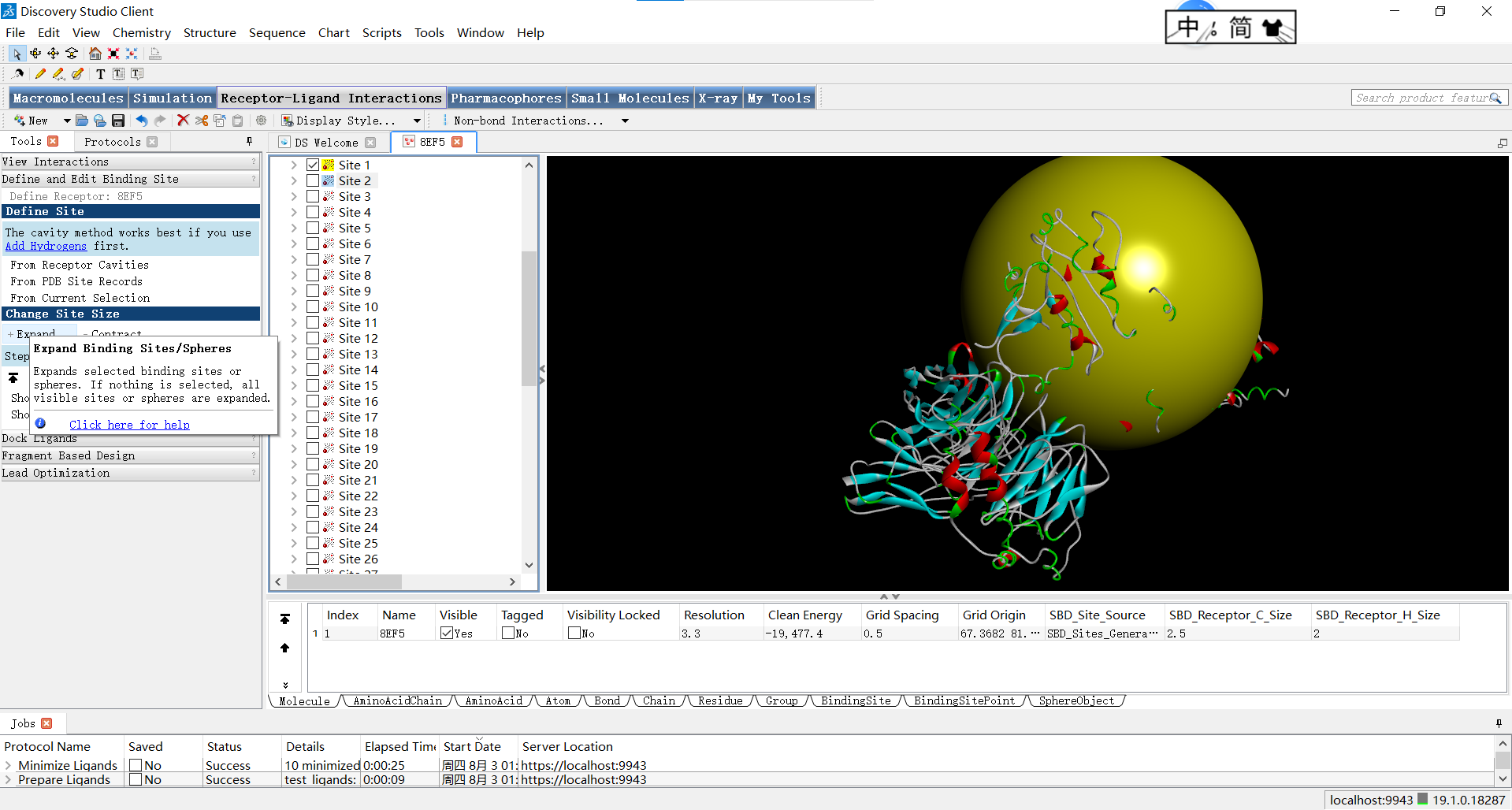
我们也可以通过另一种方式调整红球的参数:选中红球后点击右键展开菜单栏,点击attribute of 球体名称
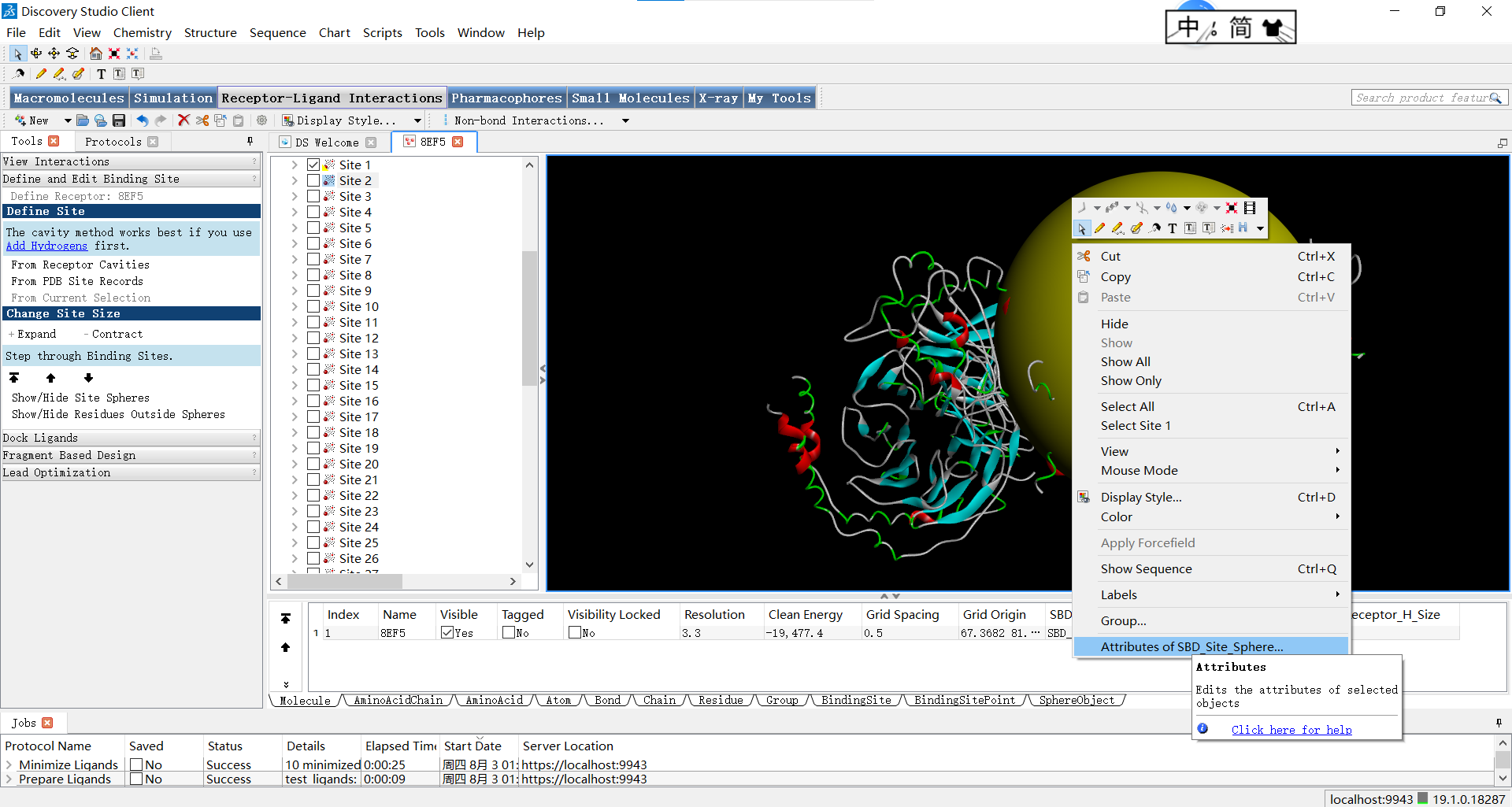
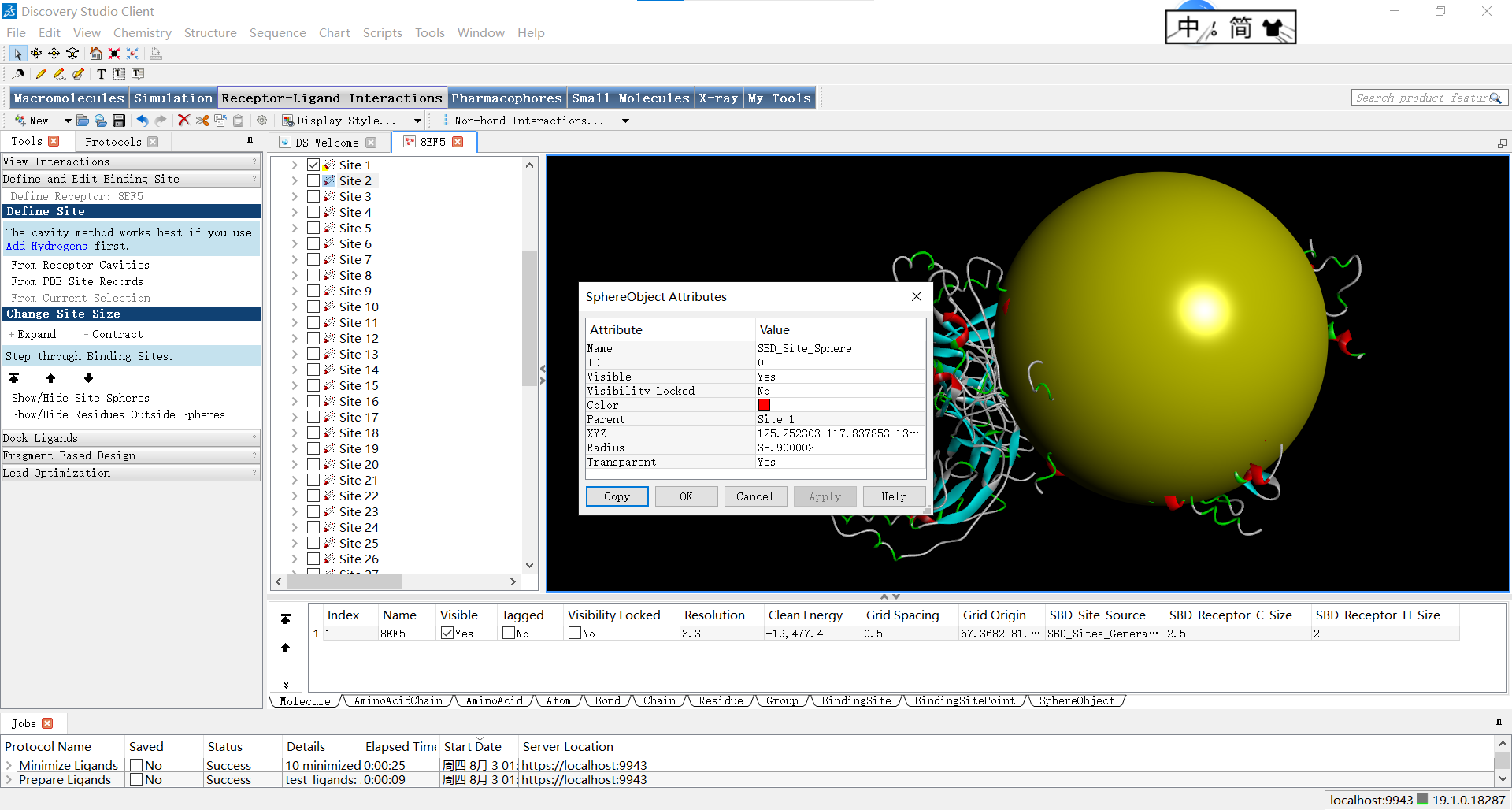
其中要注意的一点是,半径的调整会对描述蛋白质活性位点的形状特征产生影响,并且直接影响后续对接的结果,因此需要十分的谨慎。它可能会导致以下几个方面的变化:
- 捕捉到的空间范围:通过调整半径,可以增加或减少描述符所捕捉到的空间范围。较小的半径将仅考虑活性位点中的更局部区域,而较大的半径将包括更广泛的周围空间。因此,选择合适的半径可以影响描述符对活性位点内外空间的表征程度。
- 形状特征的表示:调整半径还会对活性位点的形状特征进行表示。较小的半径可能更关注较细微的形状细节,而较大的半径可能更关注整体形状和空间分布。因此,半径的调整可能会改变描述符对于活性位点形状特征的表达方式。
- 配体相互作用的涵盖程度:活性位点半径的调整也会影响描述符对与配体相互作用相关的残基或功能残基的涵盖程度。较小的半径可能只包含位于活性位点核心的关键残基,而较大的半径可能还包括邻近残基。因此,调整半径可以影响描述符对配体相互作用特征的表示。
- 计算效率和精确性的权衡:选择合适的半径还需要平衡计算效率和描述符精确性之间的关系。较小的半径可能导致复杂的计算和较长的计算时间,而较大的半径可能会减少计算量但会丧失一些细节信息。因此,半径的调整需要考虑计算资源和应用需求之间的平衡。
因此,小伙伴们在进行 SBD_Site_Sphere 的半径调整时,有几个注意事项需要考虑:
- 根据目标蛋白质的特性进行调整:SBD_Site_Sphere 的半径应该根据目标蛋白质的结构和功能进行调整。不同的蛋白质可能具有不同大小和形状的活性位点,因此需要根据具体情况进行调整。
- 考虑与配体的相互作用:活性位点的半径应该适当地包含与配体相互作用的关键残基或功能残基。这样可以确保活性位点的描述符能够捕捉到蛋白质和配体之间的相互作用特征。
- 平衡精确性和计算效率:调整半径时需要平衡描述符的精确性和计算效率之间的关系。较小的半径可以提供更精确的描述,但会增加计算复杂性。较大的半径可以减少计算量,但可能会损失一些细节信息。需要根据具体需求进行权衡选择。
- 通过交叉验证进行验证:为了确认所选的半径调整是否合适,可以使用交叉验证等方法进行验证。将调整后的 SBD_Site_Sphere 描述符应用于一组已知的蛋白质-配体复合物,然后评估其预测性能和准确性。
小果在这里建议初学的小伙伴们,如果没有十分确定的合适参数,保持软件默认的参数即可,可以通过change site size下的contract和expand进行微调,轻易不要大幅更改半径参数。
分子对接如何操作?
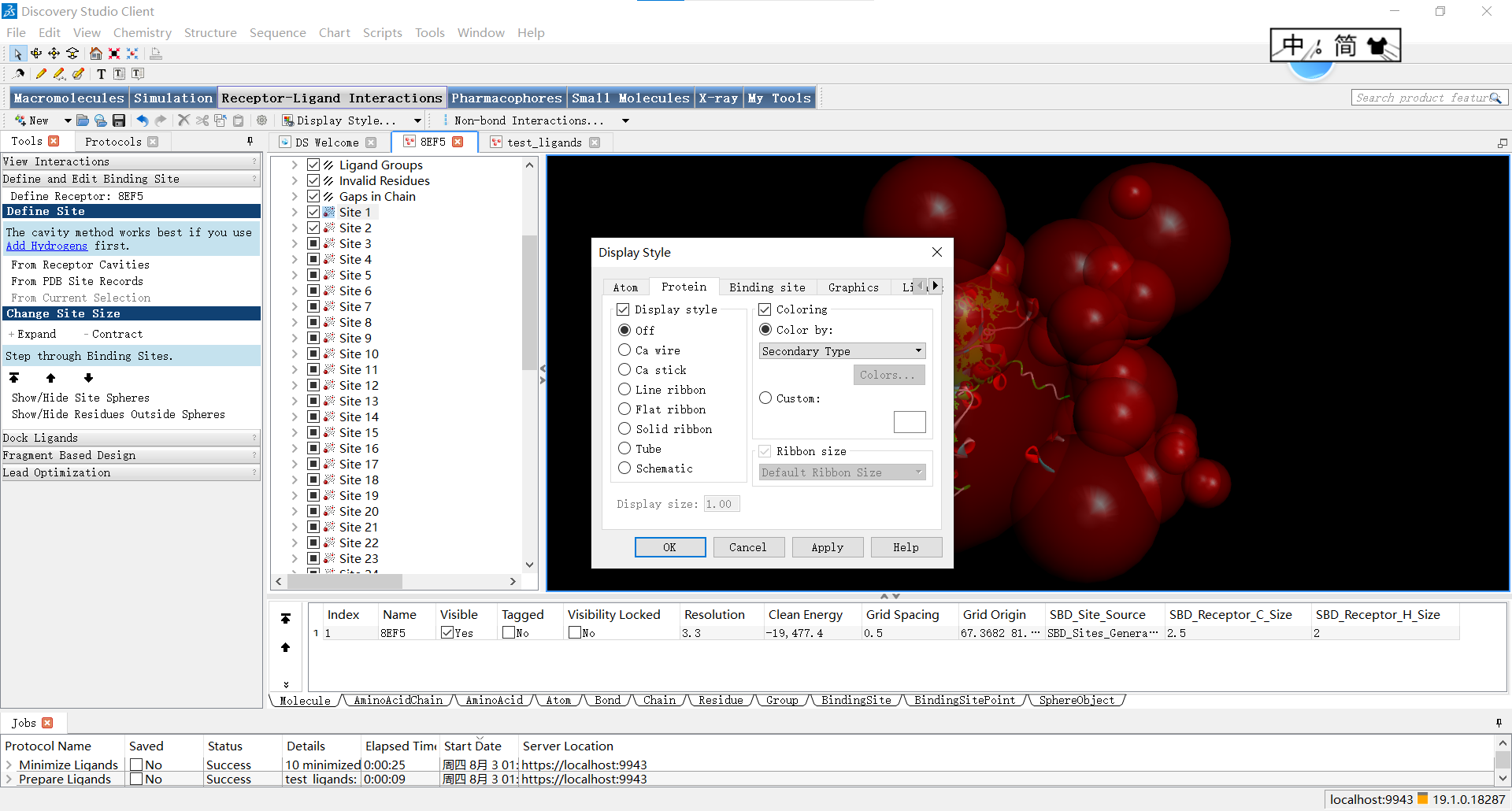
首先,为了便于观察蛋白,小果习惯第一步先让蛋白线性显示,具体调整方式如下:在黑色区域右键,点击display style
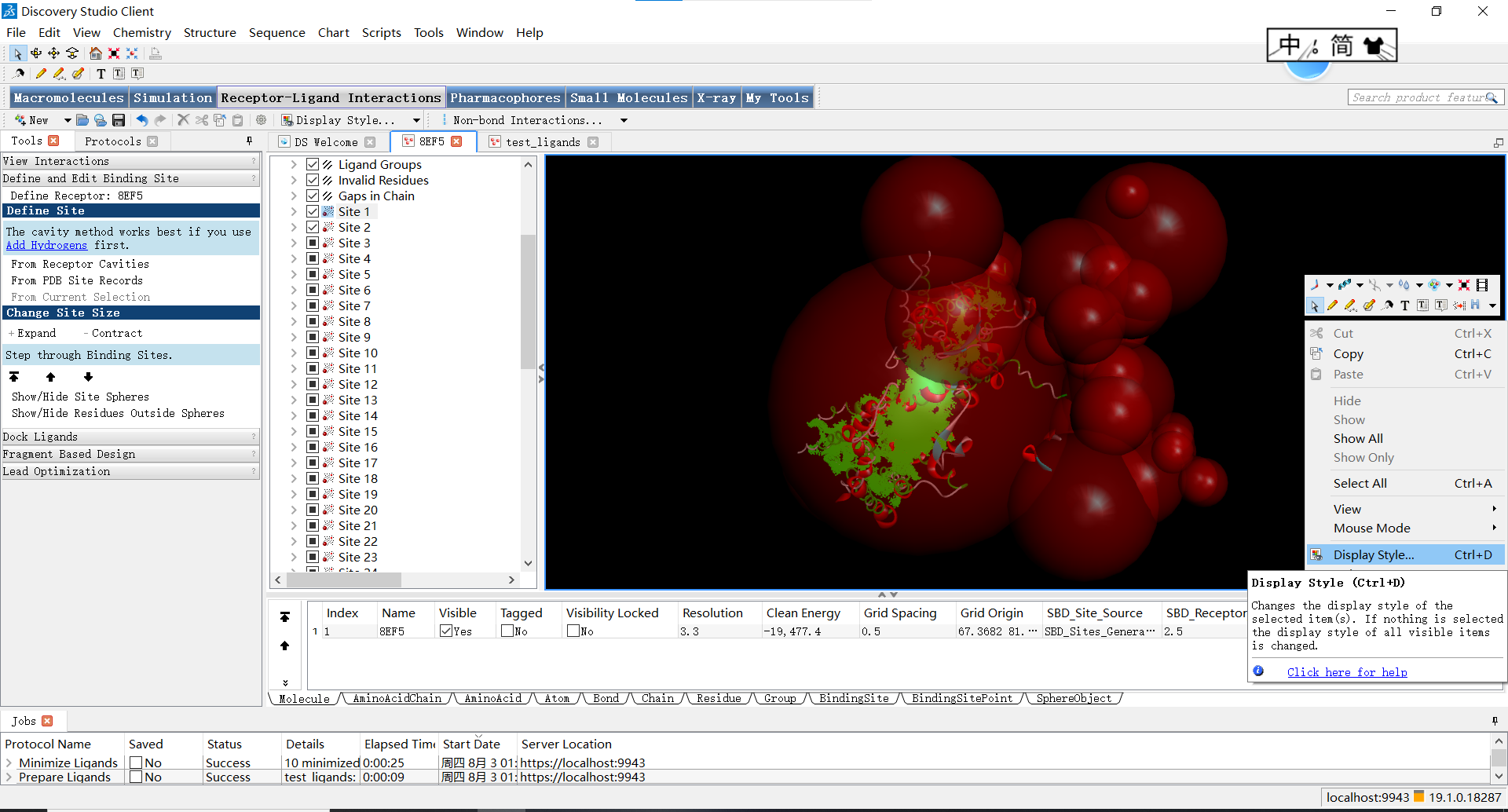
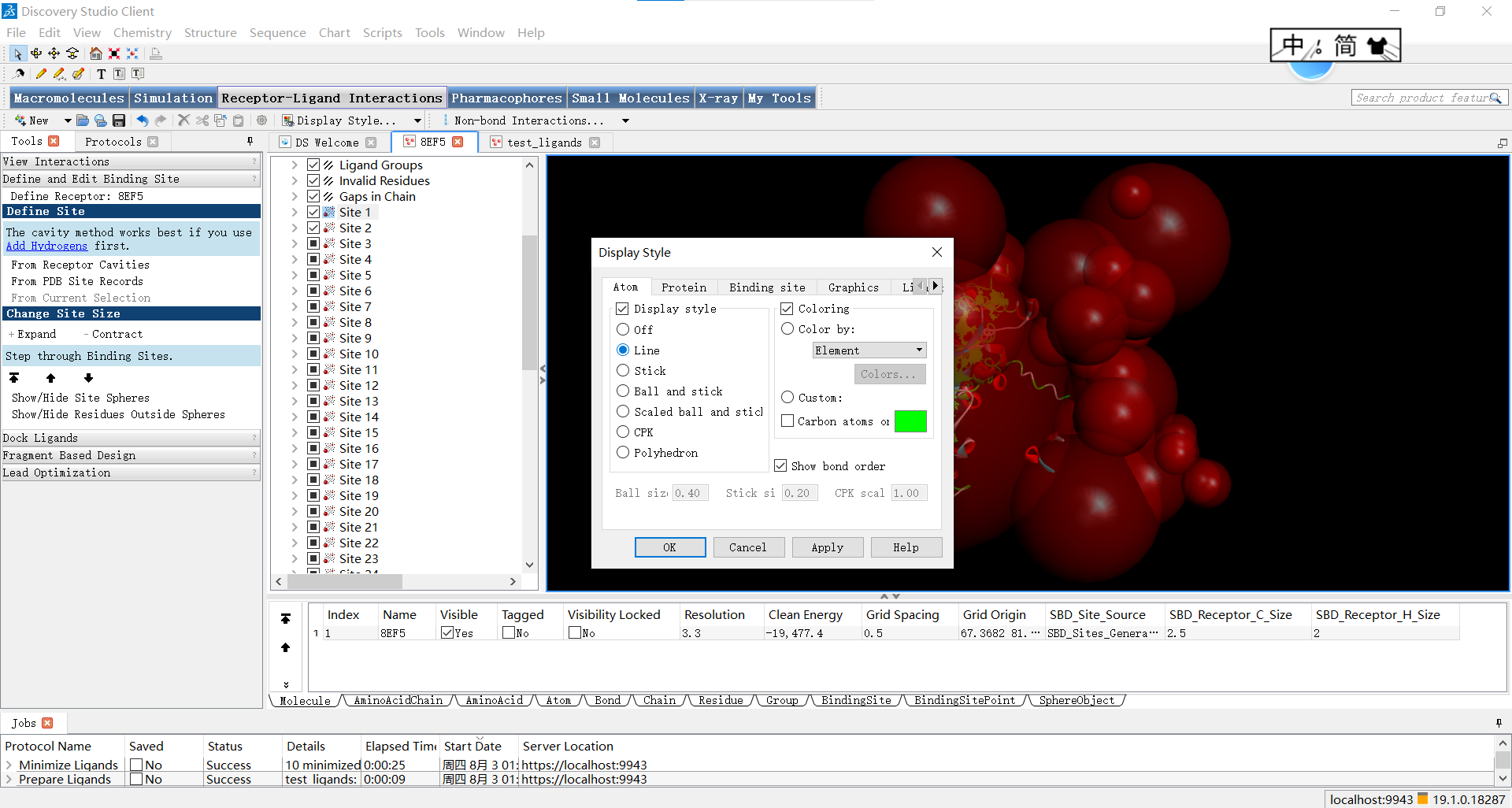
随后关闭蛋白的显示并让原子以线性方式显示:
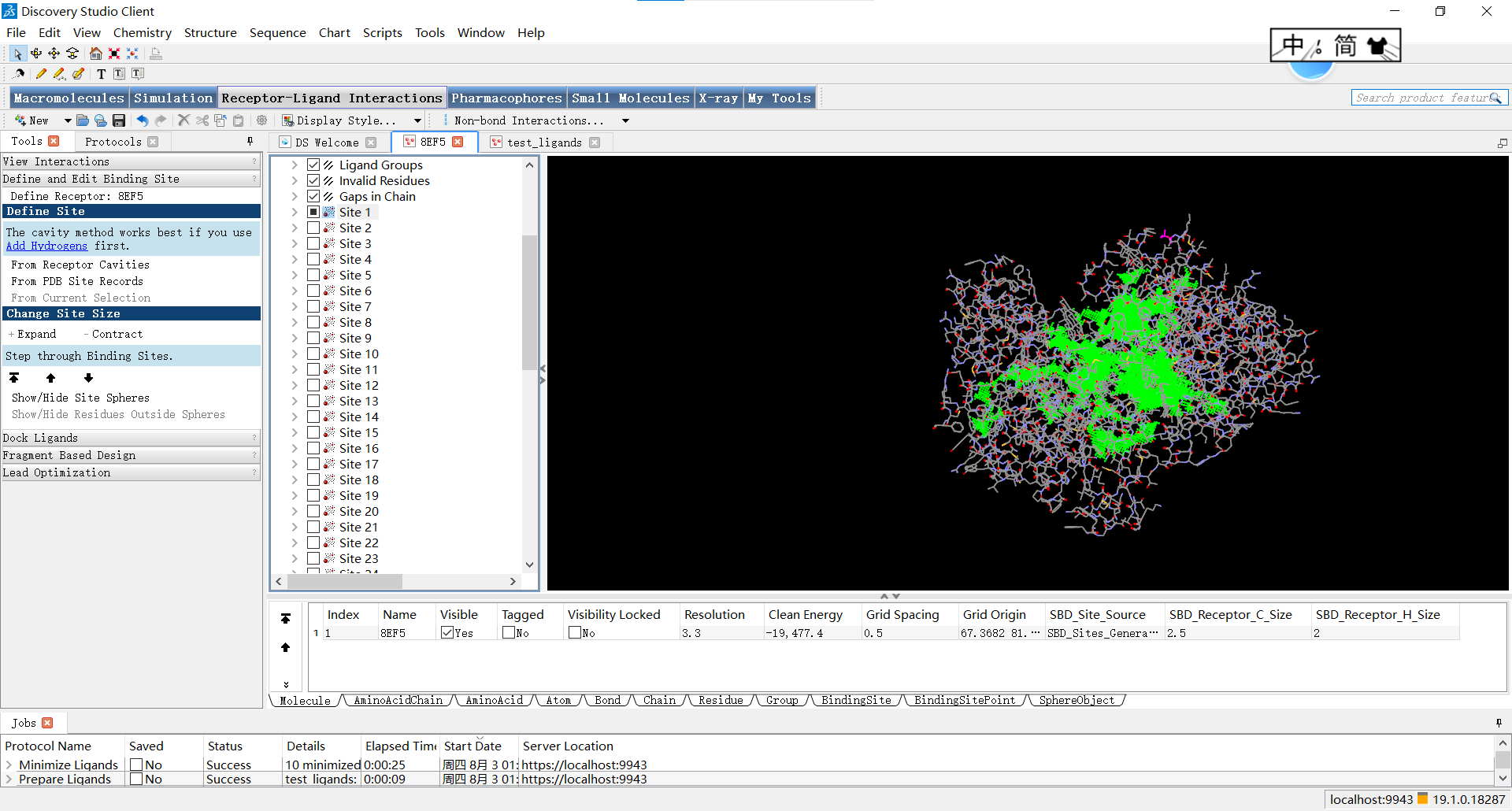
再关闭红球的显示
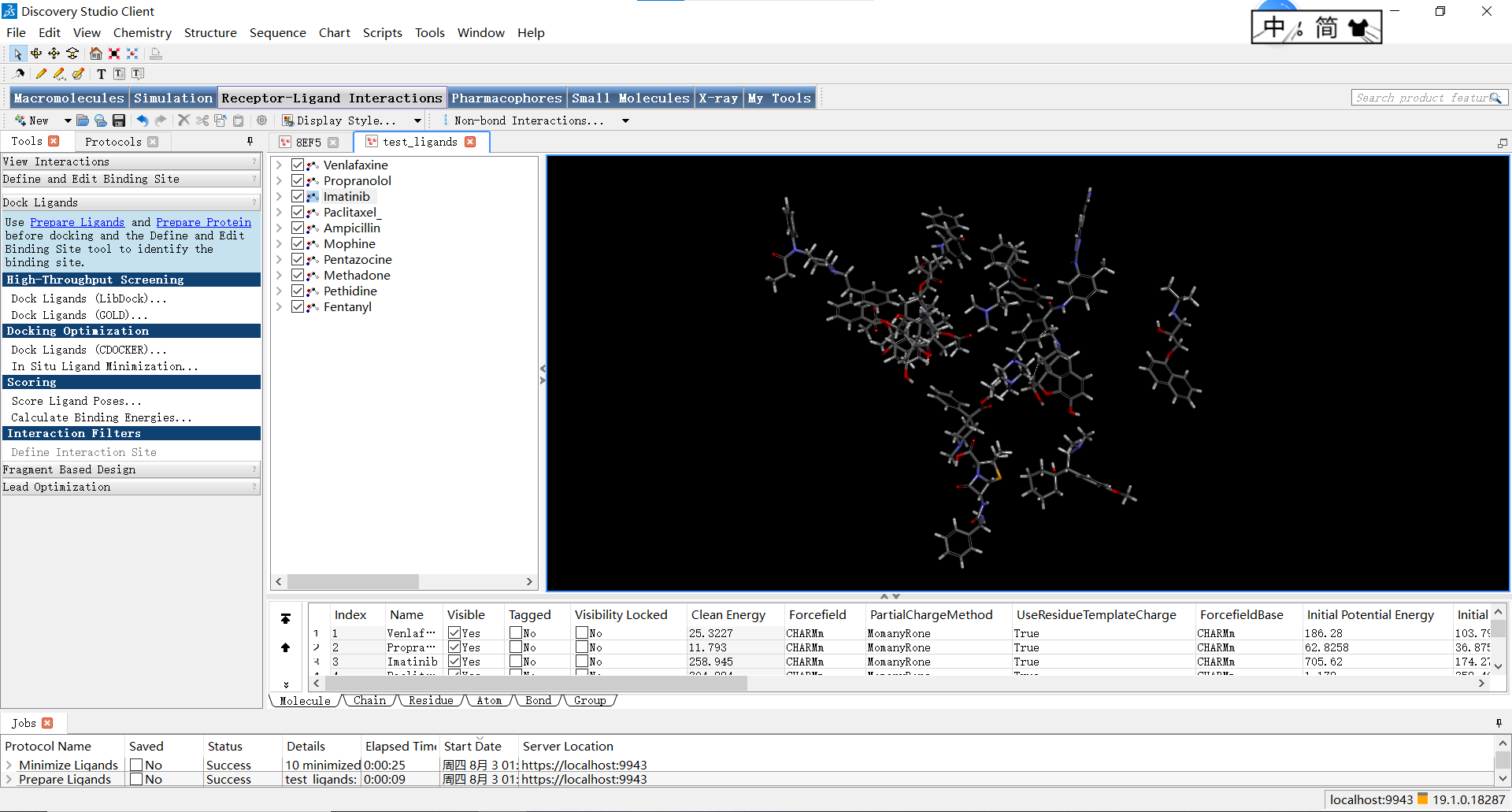
随后我们打开处理好的小分子配体
然后就可以点开分子对接选项啦:Receptor-Ligand Interactions->Dock Ligands,可以看到这里有三种方式:Dock Ligands (LibDock)、Dock Ligands (GOLD)、Dock Ligands (CDOCKER)
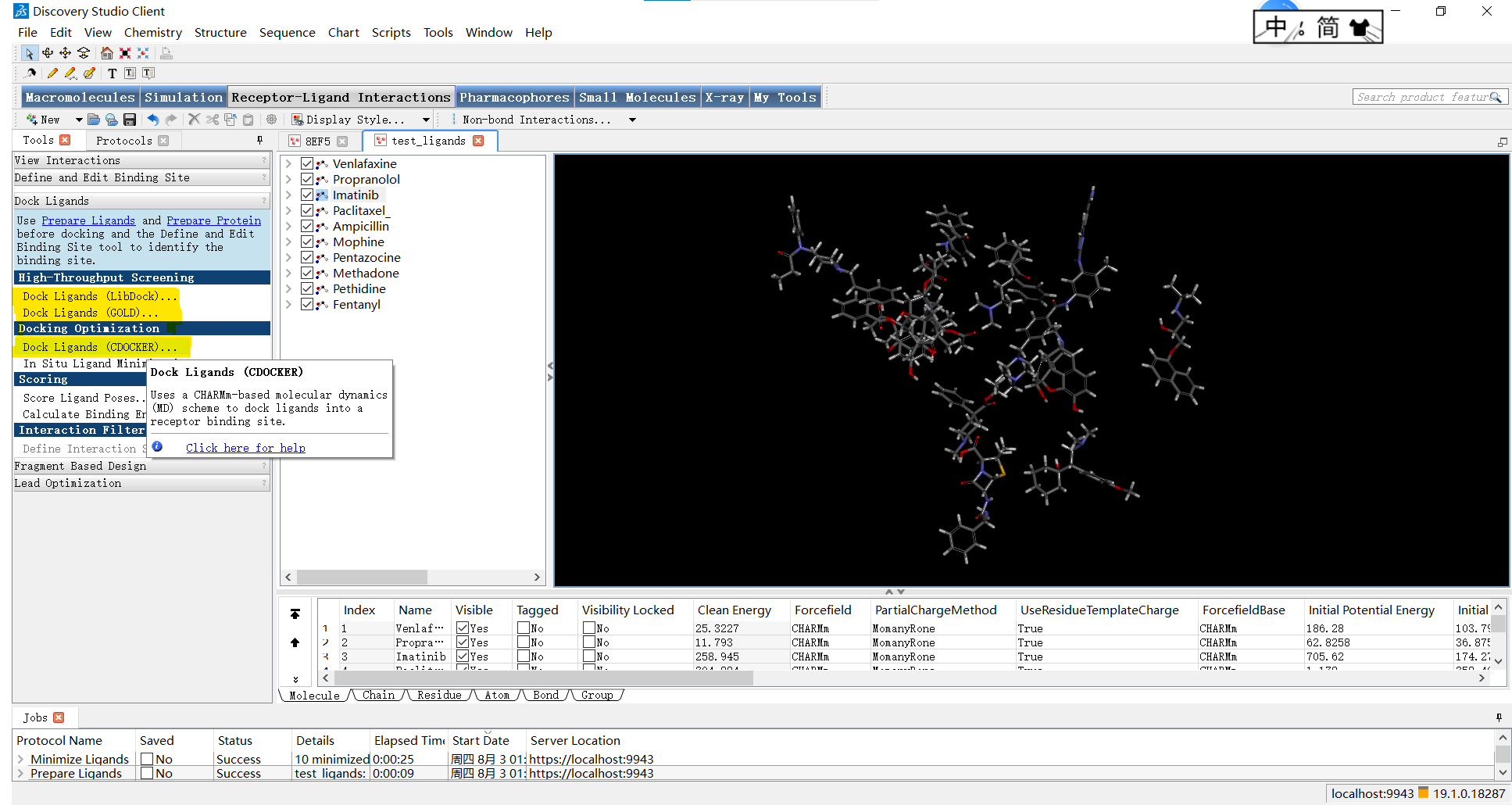
小果在这里给大家简单介绍一下,”LibDock”、”GOLD”和”CDOCKER”是三种分子对接软件,它们的介绍如下:
- “LibDock”是一种常用的基于晶格匹配和特征评分的对接方法,它使用小分子库进行高通量筛选,通过判断分子与蛋白质之间的亲和力来预测结合能力和亲合性。
- “GOLD”(Genetic Optimization for Ligand Docking)是一种基于遗传算法的对接程序,它通过在分子内搜索合适的位姿和构象来寻找最佳的配体-蛋白质结合方式。”GOLD”使用分子力学力场和评分函数进行评估,并利用遗传算法进行优化。
- “CDOCKER”(Chemical Docking)是一种基于药物设计的自动化对接软件,它使用分子对接算法来预测小分子与蛋白质之间的相互作用。”CDOCKER”使用的是一种基于能量和相互作用的评估指标,以确定最佳的配体-蛋白质结合模式。
这里小果再对三种分子对接软件的适用情况、特点和优劣进行简要说明:
LibDock:
- 适用情况:适用于高通量筛选,从大型小分子库中快速筛选出可能的结合配体。
- 特点:基于晶格匹配和特征评分,速度较快,适用于大规模筛选。
- 优劣:
- 优点:计算速度快,适用于高通量筛选;易于使用和实施。
- 缺点:较简化的评分函数,可能无法捕捉到复杂的相互作用;结果可能有一定误差。
GOLD:
- 适用情况:适用于预测精确的配体-蛋白质结合位姿,并优化配体的构象。
- 特点:基于遗传算法,能够搜索多种配体位姿和构象,并通过评分函数进行优化。
- 优劣:
- 优点:能够找到更准确的配体结合位姿和构象;适用于结合模式较复杂的系统。
- 缺点:计算耗时较长;需要更多的参数和设置,较复杂。
CDOCKER:
- 适用情况:适用于药物设计和药物优化过程中的自动化对接。
- 特点:基于能量和相互作用评估配体-蛋白质结合,支持分子力学模拟。
- 优劣:
- 优点:适用于药物设计过程,能够考虑能量和相互作用;能够进行分子力学模拟。
- 缺点:需要较长的计算时间;结果可能受到力场参数的影响。
感兴趣的小伙伴可以去查阅官方文档获得更详细的信息:
三种分子对接方法的操作方法对于我们使用者来说大同小异,这里小果就以CDOCKER为例向大家介绍:
点击CDOCKER,参数Input Receptor 设置为蛋白名:蛋白名;参数Input Ligands 设置为小分子名:All;Input Site Sphere参数为定义好的活性位点的坐标;
小果提醒大家,其余参数根据自己的需求设置,没有统一标准,但每一项参数的设置都应该十分谨慎,因为会对后续研究造成很大的影响。例如,Top Hits 下的Pose Cluster Radius参数(指在分子对接过程中,用于对生成的配体位姿进行聚类的半径值,位于该半径内的位姿将被认为是相似的,并被归为同一类)一般设置的会较大一些,从而减少生成的位姿数量,这有助于提高研究效率并减少后续分析的复杂性。这里小果设置为了0.5.
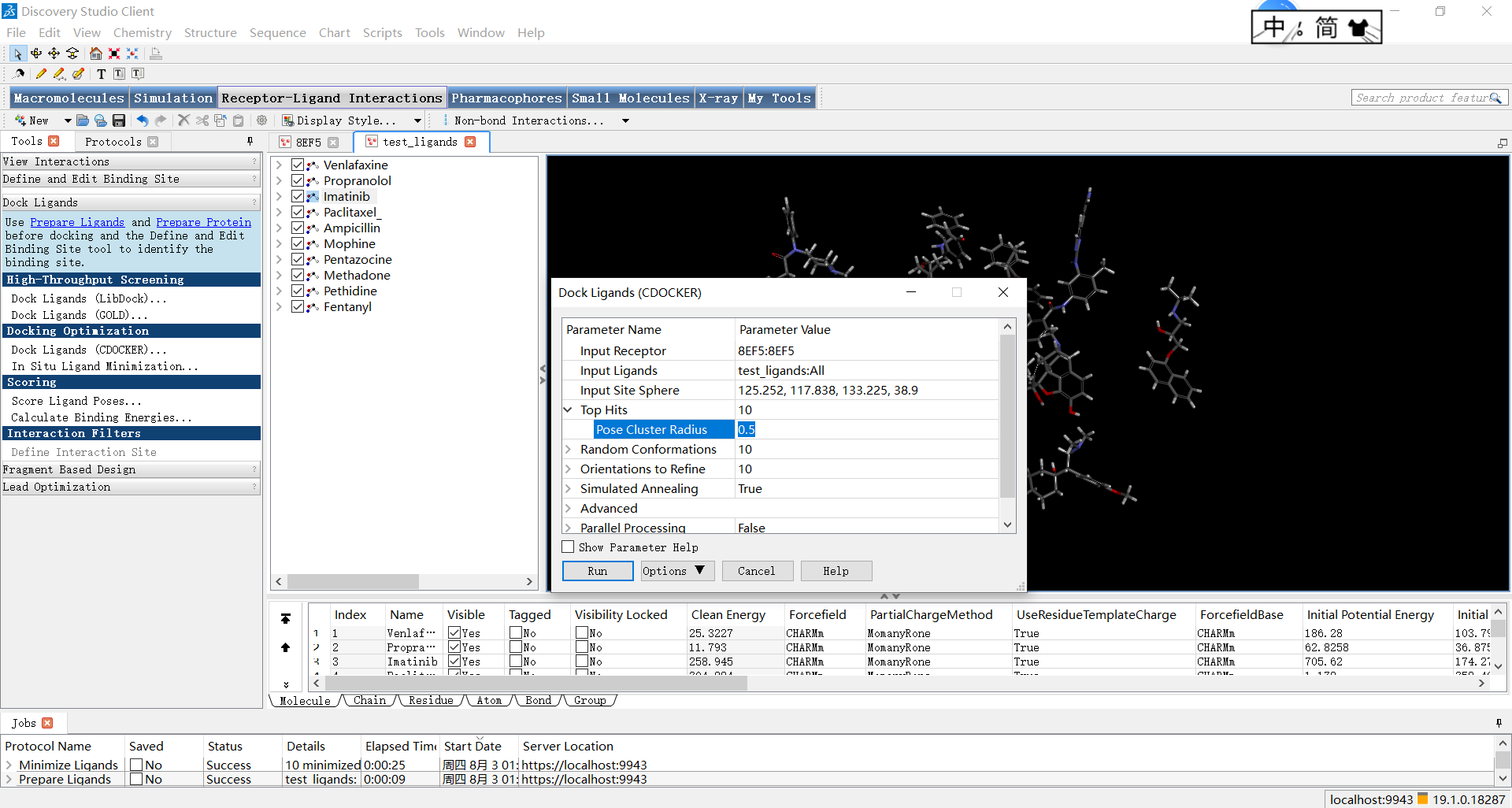
参数设置完成后,点击run等待一会即可。限于篇幅,分子对接的进阶内容和结果分析小果会在下次分享中介绍,本次的分享就到这里啦,如果小伙伴们平时在生信分析的操作过程中遇到困难,欢迎大家使用小果开发的生信工具平台http://www.biocloudservice.com/home.html哦。我们下次再见,拜拜~