大家知道基因与临床数据之间的相关性分析层出不穷,并通过可视化手段将分析结果展示出来,今天小果带给大家一个beeswarmR包, 用于在R中绘制点图,特别适用于展示小样本、分类数据和多类别数据的分布情况。
beeswarm是一个在R语言中用于绘制点图(swarm plot)的包。点图是一种可视化方法,用于展示数据点在一个维度上的分布情况,特别适用于小样本和分类数据。在点图中,数据点会根据另一个维度进行分组,而不会互相重叠,以避免数据重叠和遮挡。这使得点图可以更清晰地显示数据的分布情况和密度。
本期主要给大家带来在进行基因与临床数据的相关性分析时,通过绘制箱线图和点图的方式,将基因表达在不同临床分组下的分布情况可视化呈现出来。这有助于研究人员快速发现基因在不同临床情况下的差异性。
#install.packages(“beeswarm”)
#安装名为beeswarm的R包。
library(beeswarm)
setwd(“”) #使用setwd()函数设置工作目录为特定路径修改工作目录
expFile=”rocSigExp.txt”
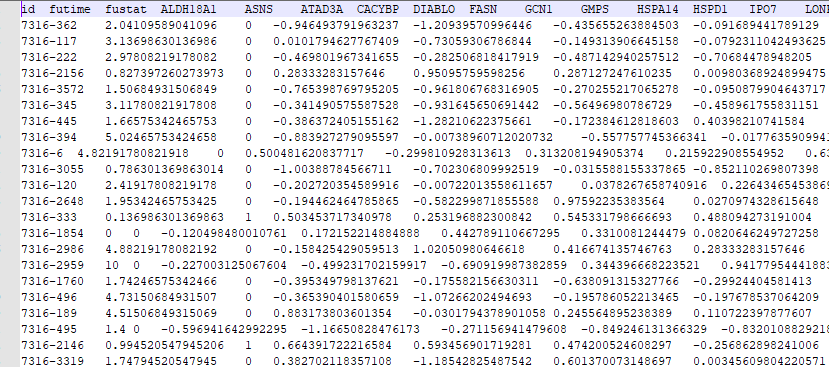
#表达数据文件
clinicalFile=”clinical.txt”
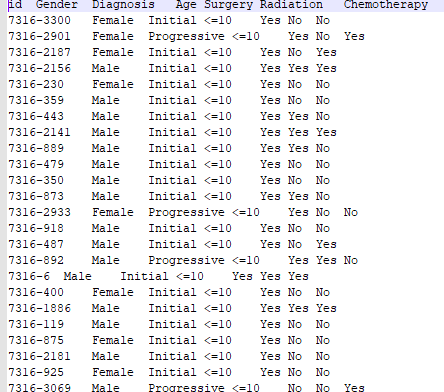
#临床数据文件
exp=read.table(expFile,sep=”\t”,header=T,check.names=F,row.names=1)
#读取表达数据文件
cli=read.table(clinicalFile,sep=”\t”,header=T,check.names=F,row.names=1)
#读取临床数据文件
exp=exp[,3:ncol(exp)]
#从表达数据中选择第3列到最后一列的数据
samSample=intersect(row.names(exp),row.names(cli))
#将两个数据集的样本ID进行交集运算,以确保表达数据和临床数据的样本一致性。
exp=exp[samSample,]
cli=cli[samSample,]
pFilter=0.05
#临床相关性分析,输出表格
outTab=c()
#初始化一个空的数据框 outTab,用于存储临床相关性分析的结果。
outTab=rbind(outTab,c(“id”,colnames(cli),”SigNum”))
colnames(outTab)=c(“id”,colnames(cli),”SigNum”)
for(i in colnames(exp)){
#循环遍历基因表达数据的每一列。
clinicalPvalVector=c()
sigSum=0
#内部循环中,对每个临床变量进行遍历分析,检验基因表达与临床变量的相关性。
for(clinical in colnames(cli)){
tab1=table(cli[,clinical])
labelNum=length(tab1)
rt1=cbind(expression=exp[,i],clinical=cli[,clinical])
#根据临床变量的不同水平,选择适当的统计检验方法(Wilcoxon秩和检验或Kruskal-Wallis检验)。
if(labelNum==2){
cliTest<-wilcox.test(expression ~ clinical, data=rt1)
}else{
cliTest<-kruskal.test(expression ~ clinical, data = rt1)}
pValue=cliTest$p.value
clinicalPvalVector=c(clinicalPvalVector,pValue)
#将p值存储在clinicalPvalVector中,并计算满足显著性条件(小于pFilter)的次数 sigSum。
if(pValue<pFilter){
sigSum=sigSum+1
#if语句,检查labelNum的值。根据labelNum的值,不同的颜色会被添加到dotCol向量。#如果labelNum是3,将额外的颜色添加到dotCol。同样,如果labelNum是4或大于4,会#使用rainbow()函数添加不同的颜色。
xlabel=vector()
dotCol=c(2,3)
if(labelNum==3){
dotCol=c(2,3,4)
}
if(labelNum==4){
dotCol=c(2,3,4,5)
}
if(labelNum>4){
dotCol=rainbow(labelNum)
}
for(j in 1:labelNum){
xlabel=c(xlabel,names(tab1[j]) )
}
#设定p值格式
pval=ifelse(pValue<0.001,”<0.001″,paste0(“=”,sprintf(“%.03f”,pValue)))
#绘制临床相关性箱线图
#用boxplot()函数绘制箱线图,显示基因表达在不同临床分组间的分布情况。
#使用beeswarm()函数绘制点图(beeswarm图),将数据点分布在箱线图附近,以更清晰地显示分布情况。
b = boxplot(expression ~ clinical, data = rt1,outline = FALSE, plot=F)
yMin=min(b$stats)
yMax = max(b$stats/5+b$stats)
#计算了箱线图中的y轴范围。b$stats是boxplot函数计算的统计信息,yMin设置为最小值,yMax设置为最大值的五分之一加上最大值。
n = ncol(b$stats)
outPdf=paste0(“cliCor.”,i,”_”,clinical,”.pdf”)
width=ifelse(clinical==”diagnosis”,8,7)
#根据clinical的值,设置PDF文件的宽度,如果clinical是”diagnosis”,宽度为8,否则为7。
pdf(file=outPdf,width = width,height = 5)
par(mar = c(4.5,6,3,3))
boxplot(expression ~ clinical, data = rt1,names=xlabel,
ylab = “Protein expression”,main=paste0(i,” (p”,pval,”)”),xlab=clinical,
cex.main=1.3, cex.lab=1.2, cex.axis=1.1,ylim=c(yMin,yMax),outline = FALSE)
beeswarm(expression ~ clinical, data = rt1, col =dotCol, lwd=0.1, pch = 16, add = TRUE, corral=”wrap”)
#绘制箱线图,其中names参数设置为之前定义的xlabel向量,y轴标签设置为”Protein expression”,主标题由i和pval组成,x轴标签设置为clinical的值,调整了标题、标签和轴的字体大小。
dev.off()
}
}
geneClinical=c(i,clinicalPvalVector,sigSum)
outTab=rbind(outTab,geneClinical)
#将每个基因与各临床变量的相关性分析结果存储在数据框 outTab 中。
#设置outTab 的列名为基因名、各临床变量名、以及一个额外的列 “SigNum”,表示显著性相关的次数。
#将 outTab 数据框写入名为 “cliCorResult.xls” 的文件,保存分析结果。
}
write.table(outTab,file=”cliCorResult.xls”,sep=”\t”,col.names=F,row.names=F,quote=F)
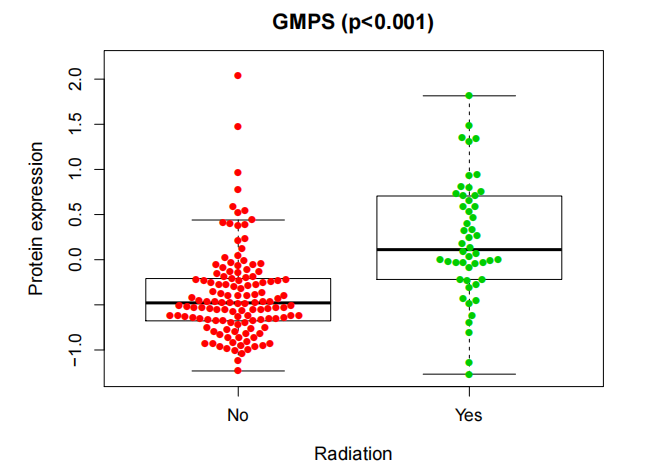
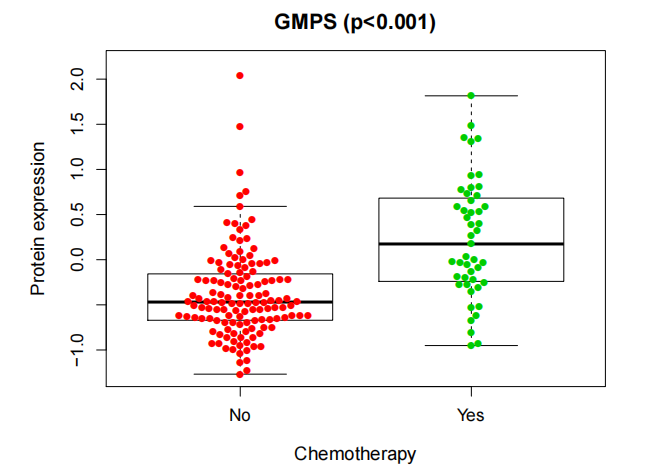
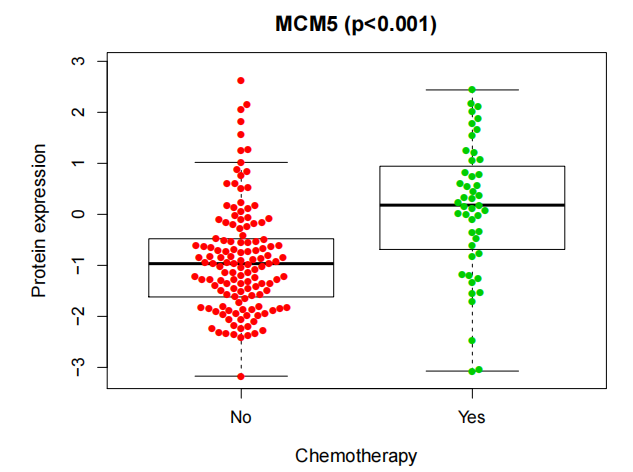
总之基因与临床数据的相关性分析,生成了箱线图和点图来展示相关性,最终将分析结果写入文件。这可以帮助研究人员理解基因与临床变量之间的关系。下期将为你带来更多R语言的骚操作技巧,以下推荐的是一个多功能的生信平台。
云生信平台链接:http://www.biocloudservice.com/home.html。