今天小果来为大家介绍细胞拟时序分析以及如何使用monocle3做细胞拟时序分析。
那么什么是细胞拟时序分析呢?
在生命体发育过程中,细胞会在特定条件下进行分化,从一个状态过渡到另一个状态,表达完全不同的基因集,有的基因被沉默,而有的基因被激活。这些状态通常很难表征,因为想要纯化中间状态的细胞很困难或不可能。但是通过单细胞测序技术捕获一群细胞的转录快照,可以使用轨迹推断(trajectory inference)的方法,对测序的细胞根据表达模式的相似性进行排序。由此一来就可以模拟细胞的动态变化。
可能有的小伙伴有疑问了,为什么叫做伪时序分析呢?
如果想要得到细胞分化轨迹,分化的时序分析,理论上的做法是设置多个时间点,每个时间点取一次样,进行测序以及后续的分析。但是这种方法很难做,而且费时费力费钱。通过单细胞测序可以从一群细胞中得到转录瞬时快照,可以从中推断出细胞的分化轨迹。这种方法因为只取了一个时间点,所以叫做伪时序分析。

Monocle3是做伪时序分析的首选软件包。其团队发表了一篇NATURE一篇NB两篇 NM。经过两个大版本的更新,monocle3还可以做细胞的降维和分类等分析。Monocle使用算法来学习每个细胞作为动态生物过程的一部分必须经历的基因表达变化序列。一旦了解了基因表达变化的总体“轨迹”,Monocle就可以将每个细胞放置在轨迹中的适当位置。然后,就可以使用Monocle的差异分析工具包来寻找在轨迹过程中受调控的基因。
接下来小果带大家用monocle3来做伪时序分析。
一、安装monocle3
首先安装Bioconductor, 打开Rstudio并且运行
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(version = “3.14”)
接着安装一些Bioconductor不会自动安装的依赖
BiocManager::install(c(‘BiocGenerics’, ‘DelayedArray’, ‘DelayedMatrixStats’,
‘limma’, ‘lme4’, ‘S4Vectors’, ‘SingleCellExperiment’,
‘SummarizedExperiment’, ‘batchelor’, ‘HDF5Array’,
‘terra’, ‘ggrastr’))
然后安装monocle3
install.packages(“devtools”)
devtools::install_github(‘cole-trapnell-lab/monocle3’)
通常这会需要一定的时间。
加载monocle3
library(monocle3)
二、数据加载与预处理
小果这里使用一个秀丽隐杆线虫数据集,这一数据集来自Packer&Zhu等人。他们的研究包括对整个胚胎发育过程的时间序列分析。我们将使用数据的一小部分,其中包括大多数神经元细胞。
#加载数据
expression_matrix <- readRDS(url(“https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_expression.rds”))
cell_metadata <- readRDS(url(“https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_colData.rds”))
gene_annotation <- readRDS(url(“https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_rowData.rds”))
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)
#数据预处理
#数据预处理
cds <- preprocess_cds(cds, num_dim = 50)
cds <- align_cds(cds, alignment_group = “batch”, residual_model_formula_str = “~ bg.300.loading + bg.400.loading + bg.500.1.loading + bg.500.2.loading + bg.r17.loading + bg.b01.loading + bg.b02.loading”)
使用alignment_group对其不同批次细胞。bg.300等每一列对应与细胞可能被污染的信号,将这些项通过residual_model_formula_str传递给align_cds,在后续的处理中减去这些背景信号。
降低维度并可视化结果
接下来可以对数据进行降维,monocle3建议使用默认的UMAP算法降维。
cds <- reduce_dimension(cds)
plot_cells(cds, label_groups_by_cluster=FALSE, color_cells_by = “cell.type”)

Monocle重建了带有许多分支的轨迹。
使用plot_cells()来可视化单个基因如何沿轨迹变化。让我们来看看一些在纤毛神经元中具有有趣表达模式的基因:
ciliated_genes <- c(“che-1”,
“hlh-17”,
“nhr-6”,
“dmd-6”,
“ceh-36”,
“ham-1”)
plot_cells(cds,
genes=ciliated_genes,
label_cell_groups=FALSE,
show_trajectory_graph=FALSE)

6个基因的轨迹变化。
聚类细胞
虽然细胞可以从一种状态持续过渡到下一种状态,它们之间没有离散的边界,但是Monocle并不认为数据集中的所有细胞都来自一个共同的转录“祖先”。Monocle能够通过其聚类程序了解细胞何时应该放置在相同的轨迹中,而不是放置在不同的轨迹中。
cds <- cluster_cells(cds)
plot_cells(cds, color_cells_by = “partition”)
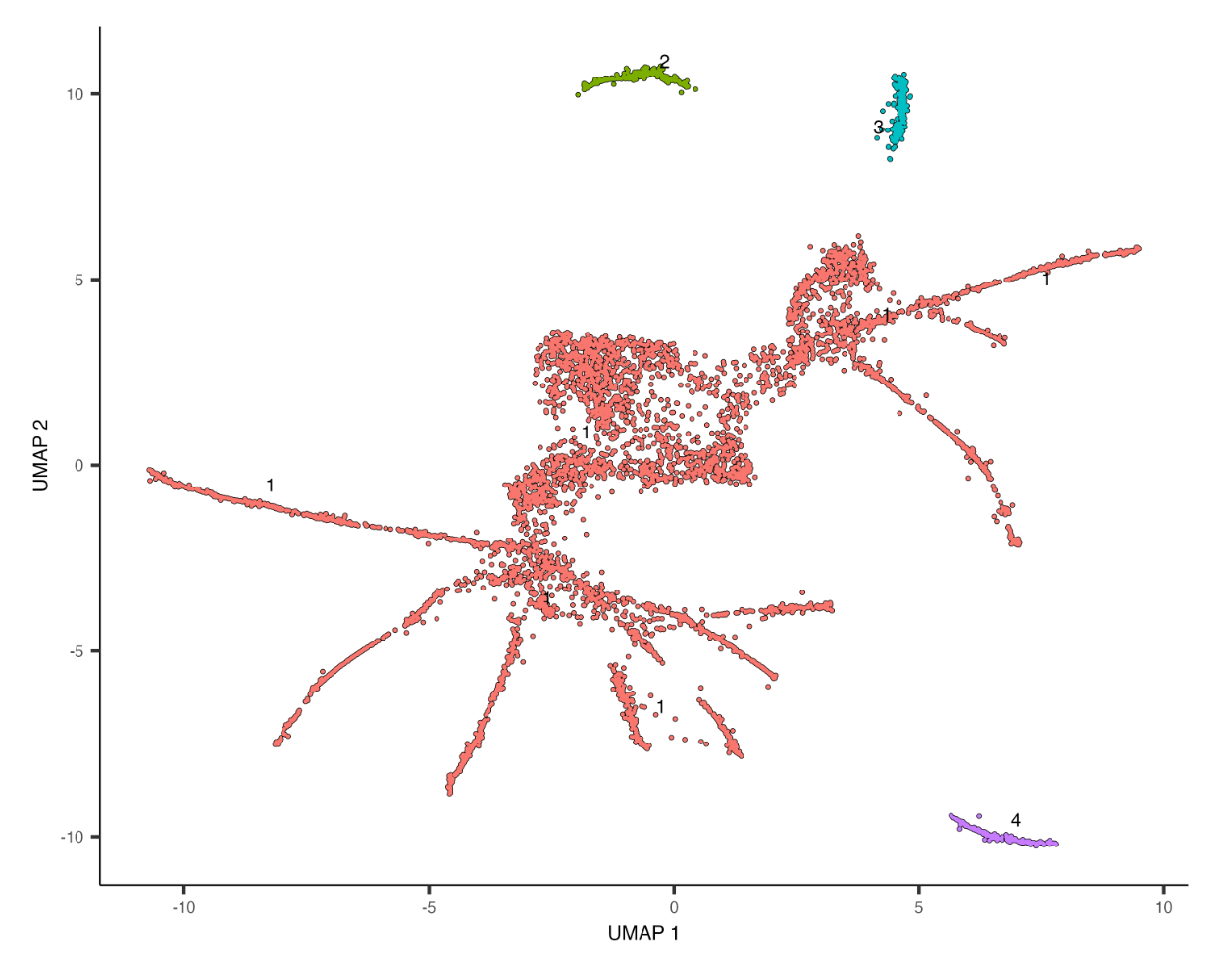
将细胞分为了四类。
接下来,我们将使用最关键的Learn_graph()函数在每个分区中拟合一个主要图:
cds <- learn_graph(cds)
plot_cells(cds,
color_cells_by = “cell.type”,
label_groups_by_cluster=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE)
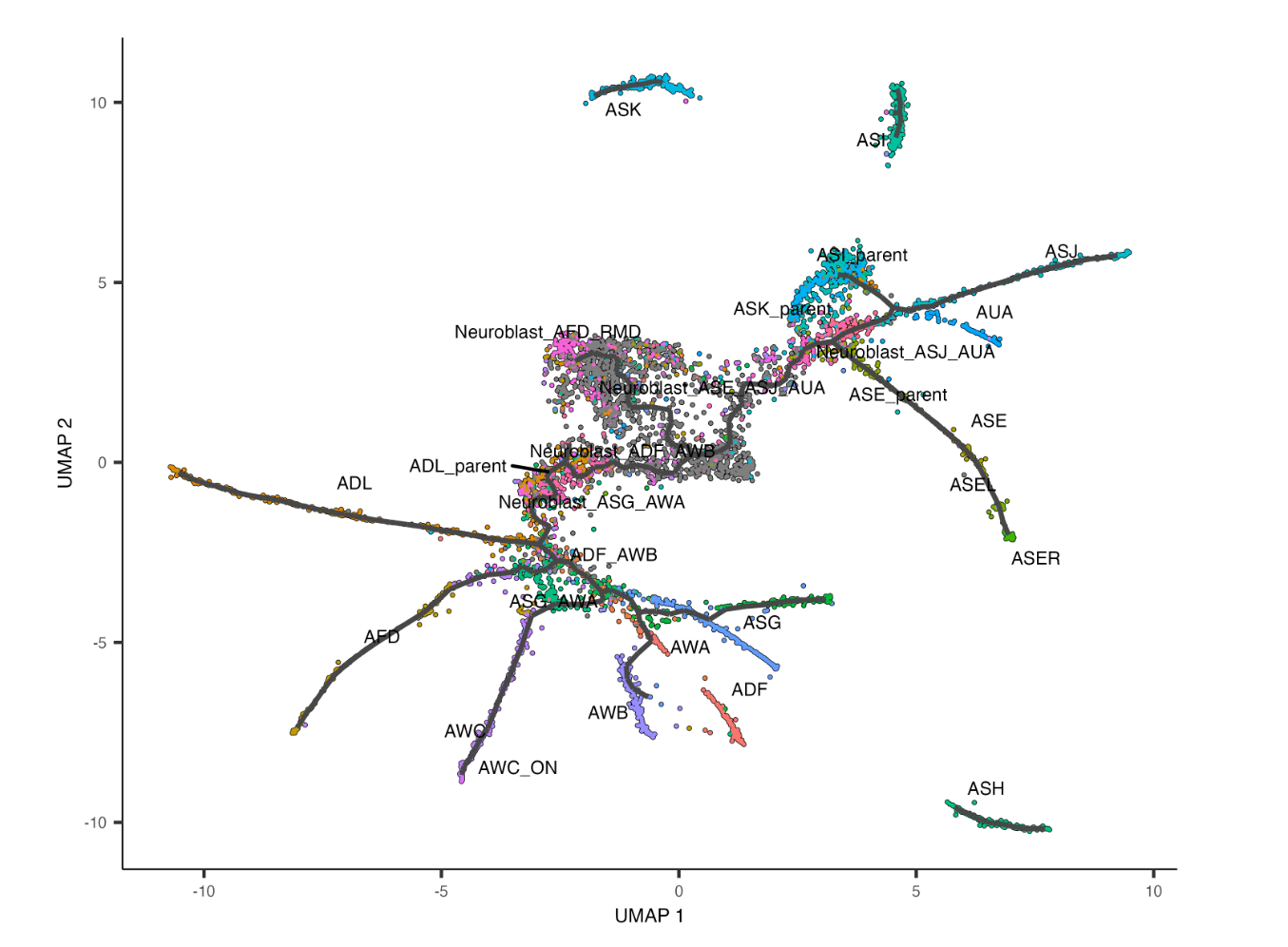
该图将用于许多下游步骤,如分支分析和微分表达式。
细胞伪时序分析
为了将细胞排列整齐,我们需要告诉Monocle生物过程的“开始”在哪里。我们通过选择图中标记为轨迹“根”的区域来做到这一点。在时间序列实验中,这通常可以通过在UMAP空间中找到由早期时间点的细胞占据的点来实现:
plot_cells(cds,
color_cells_by = “embryo.time.bin”,
label_cell_groups=FALSE,
label_leaves=TRUE,
label_branch_points=TRUE,
graph_label_size=1.5)
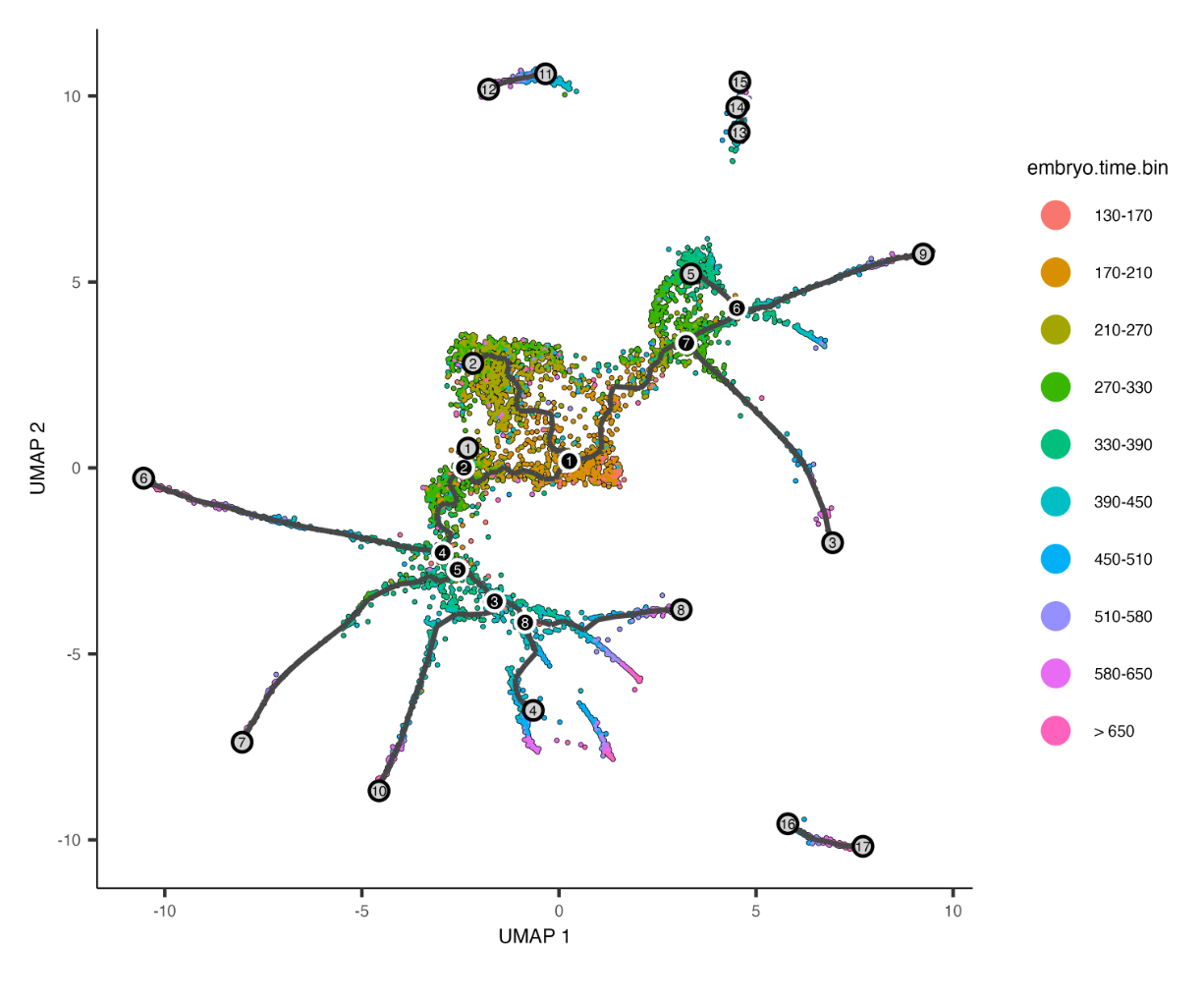
黑线显示了图的结构,带数字的圆圈表示图形中的特殊点。每片叶子,用浅灰色圆圈表示,对应于轨迹的不同结果(即细胞命运)。黑色圆圈表示分支节点,其中细胞可以传播到几个结果之一。
(一)手动选取根细胞
cds <- order_cells(cds)
现在我们已经知道了早期细胞的下落,我们可以调用order_cells(),它将计算每个细胞在伪时间内的下落。为了这样做,order_cells()需要您指定轨迹图的根节点。如果您不提供它们作为参数,它将启动用于选择一个或多个根节点的图形用户界面。
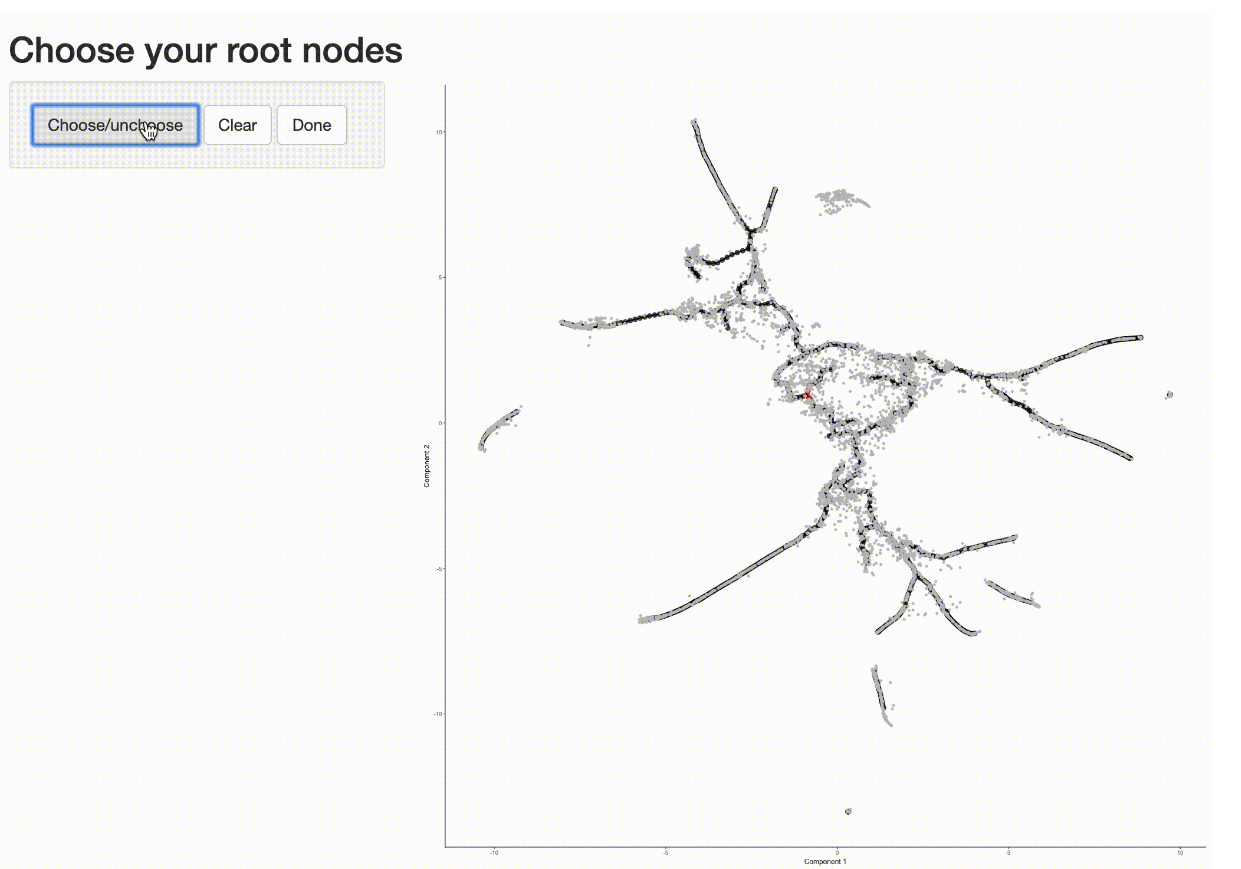
绘制细胞并通过假时间对其着色显示了它们是如何排列的:
plot_cells(cds,
color_cells_by = “pseudotime”,
label_cell_groups=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE,
graph_label_size=1.5)
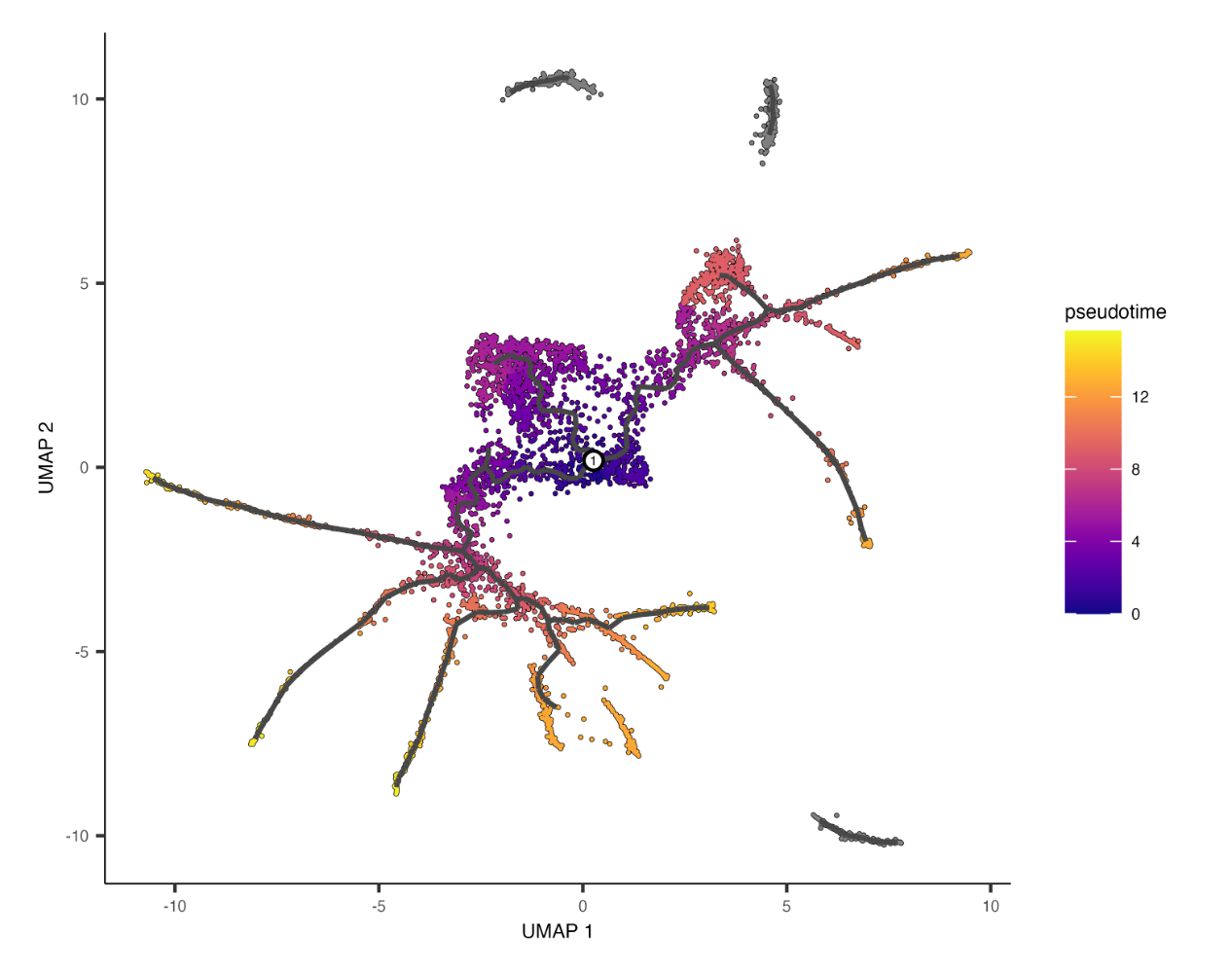
有些单元格是灰色的。这意味着它们有无限的伪时间,因为它们无法从根节点访问。
(二)通过代码拾取根节点
通常需要以编程方式指定轨迹的根,而不是手动拾取。下面的函数首先根据最接近的轨迹图节点对单元格进行分组。然后,它计算每个节点的单元格中来自最早时间点的部分。然后,它选择早期细胞占用最多的节点,并将其作为根返回。
get_earliest_principal_node <- function(cds, time_bin=”130-170″){
cell_ids <- which(colData(cds)[, “embryo.time.bin”] == time_bin)
closest_vertex <-
cds@principal_graph_aux[[“UMAP”]]$pr_graph_cell_proj_closest_vertex
closest_vertex <- as.matrix(closest_vertex[colnames(cds), ])
root_pr_nodes <-
igraph::V(principal_graph(cds)[[“UMAP”]])$name[as.numeric(names
(which.max(table(closest_vertex[cell_ids,]))))]
root_pr_nodes
}
通过root_pr_node参数将程序选择的根节点传递给order_cells()会产生
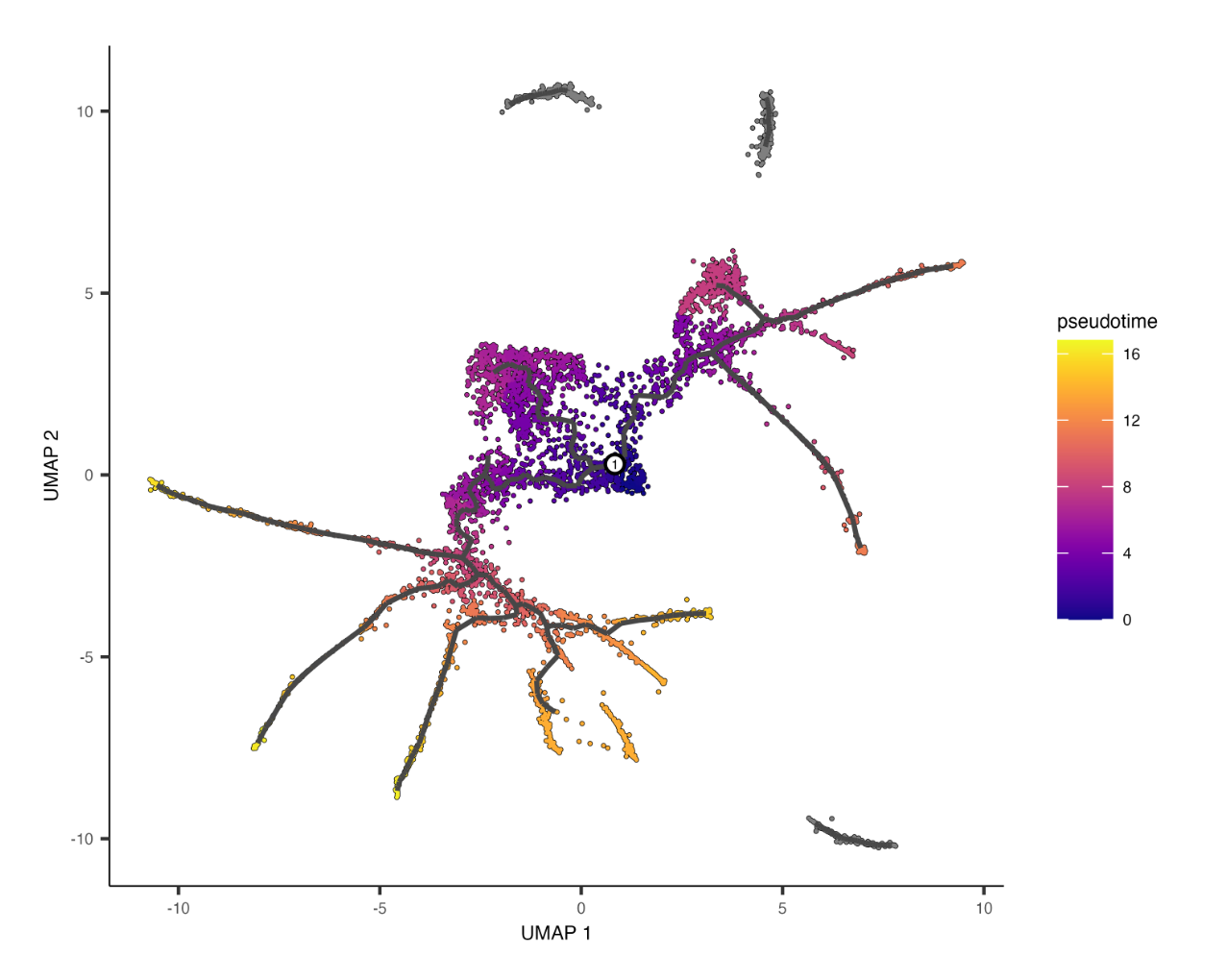
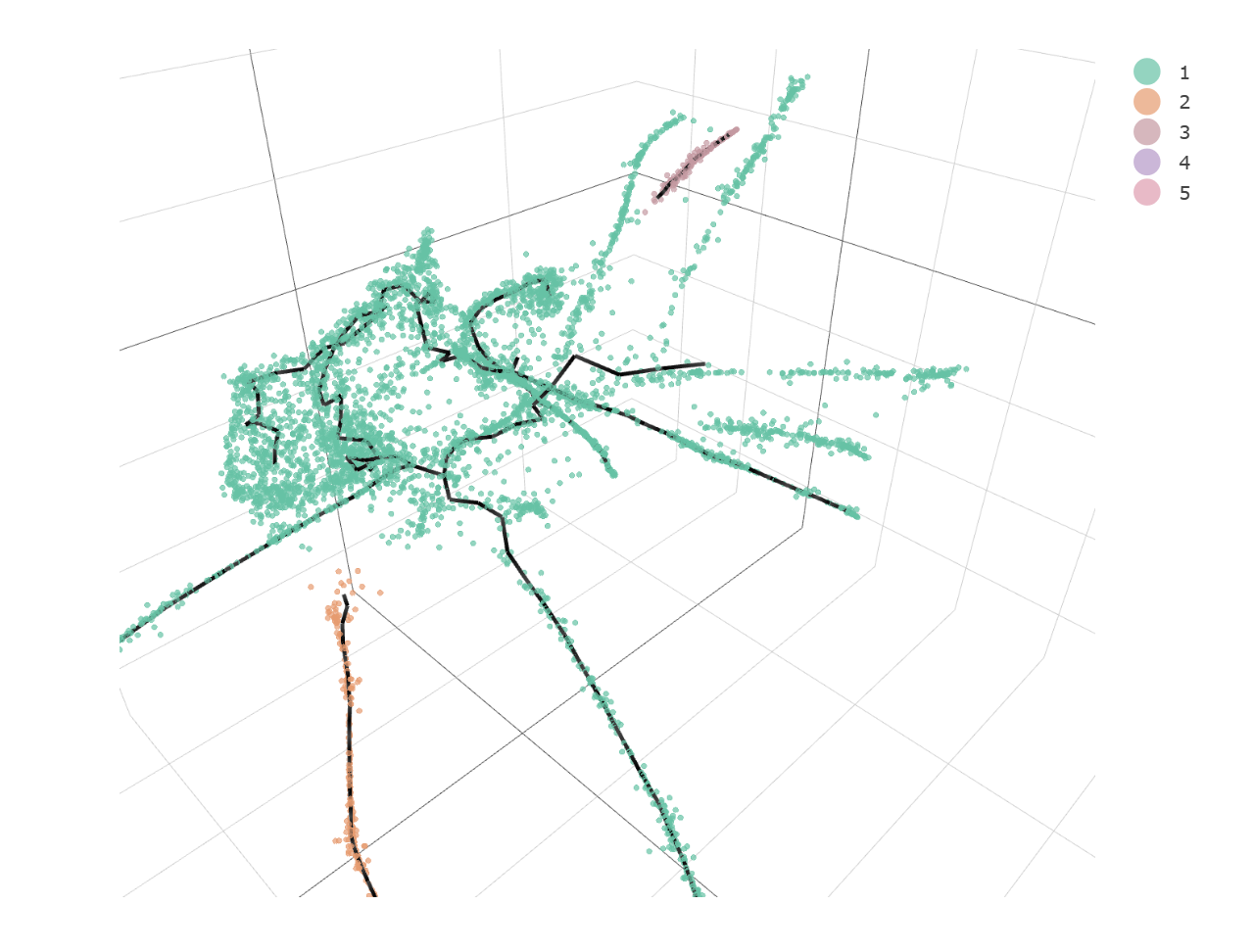
生成3D图
我们还可以生成3D的细胞分化轨迹图。
cds_3d <- reduce_dimension(cds, max_components = 3)
cds_3d <- cluster_cells(cds_3d)
cds_3d <- learn_graph(cds_3d)
cds_3d <- order_cells(cds_3d, root_pr_nodes=get_earliest_principal_node(cds))
cds_3d_plot_obj <- plot_cells_3d(cds_3d, color_cells_by=”partition”)
小果今天的讲解就到这里了~我们下期见。
本公司开发的云平台生物信息分析工具,零代码完成分析,同时也有许多生物处理的好用工具哦~云平台网址:http://www.biocloudservice.com/home.html