小伙伴还在为免疫细胞浸润分析而困扰吗,是否不知道如何去做免疫浸润分析,有时候面对单样本的基因集却不知道如何分析,今天小果给小伙伴带来一种常用于免疫细胞浸润分析的方法-ssGSEA也就是单样本GSEA。
这种方法呢是通过每个样本的基因表达数据与特定的基因集进行比较,也就是免疫细胞基因集。去估计该基因集在该样本中的相对富集程度。所以呢我们在免疫细胞分析的时候去使用ssGSEA去估计每个样本中不同免疫细胞类型的相对丰度。那么接下来就跟着小果去学习吧:
我们这里使用R代码去实现,简单高效!!
我们首先载入数据
这里呢小果使用示例数据GSE112996,小伙伴可以下载自己的GEO数据去分析。
rm(list=ls())
读入数据,这里小果准备了两个文件GSE112996_merged_fpkm_table.txt.gz和GSE112996_series_matrix.txt’
a <- read.table(‘GSE112996_merged_fpkm_table.txt.gz’,
header = T,
row.names=1)
raw_data<- a[,-1]
###表型信息提取
pheno <- read.csv(file = ‘GSE112996_series_matrix.txt’)
pheno <- data.frame(num1 = strsplit(as.character(pheno[42,]),split=’\t’)[[1]][-1],
num2 = gsub(‘patient: No.’,’P’,strsplit(as.character(pheno[51,]),split=’\t’)[[1]][-1]))
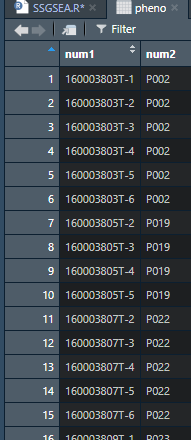
接下里我们对数据进行处理,过滤去除重复的基因名等
{
####数据过滤
data<- a[!apply(raw_data,1,sum)==0,]
####去除重复基因名的行,归一化
data$median=apply(data[,-1],1,median)
data=data[order(data$GeneName,data$median,decreasing = T),]
data=data[!duplicated(data$GeneName),]
rownames(data)=data$GeneName
uni_matrix <- data[,grep(‘\\d+’,colnames(data))]
uni_matrix <- log2(uni_matrix+1)
colnames(uni_matrix)<- gsub(‘X’,”,gsub(‘\\.’,’\\-‘,colnames(uni_matrix)))
uni_matrix<- uni_matrix[,order(colnames(uni_matrix))]
}
得到如下矩阵
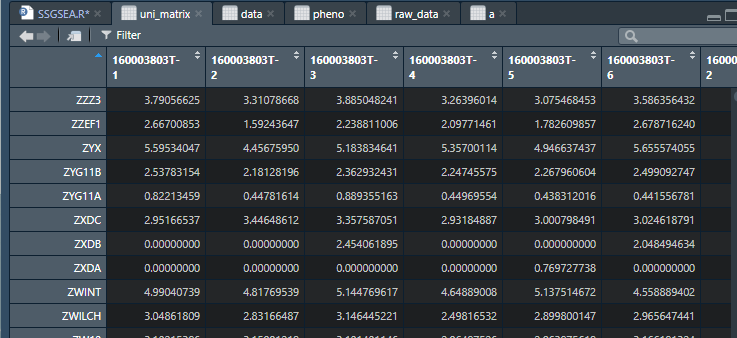
save(uni_matrix,pheno,file = ‘uni_matrix.Rdata’)#保存下来,后续使用
我们处理好之后就开始后续的分析吧
这个需要加载几个包
rm(list=ls())
##加载包
{
library(genefilter)
library(GSVA)
library(Biobase)
library(stringr)
}
小伙伴没有的包不要忘记去下载,如何下载不了就用#BiocManager::install(“genefilter”)
然后我们##载入测试数据,重点就是上述图片中两组数据
load(‘uni_matrix.Rdata’)
下面我们读取基因列表,得到免疫细胞对于的特异的基因
这里小果给大家在官网下来好,有想要的小伙伴可以下面连接获取 mmc3.csv文件
gene_set<- read.csv(‘mmc3.csv’,header = T)##读取已经下载好的免疫细胞和对应基因列表,
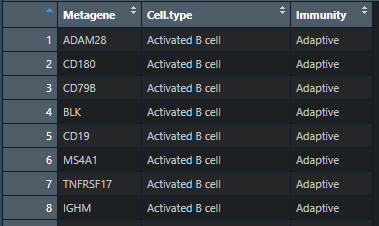
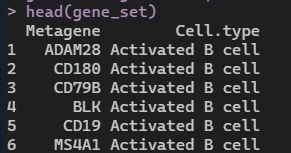
gene_set<-gene_set[, 1:2]#选取特异基因和对应的免疫细胞两行
head(gene_set)
list<- split(as.matrix(gene_set)[,1], gene_set[,2])
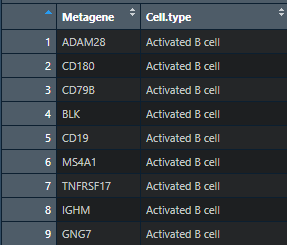 得到每种免疫细胞对于的基因
得到每种免疫细胞对于的基因
接下来我们去评估EstimatesGSVAenrichment scores 得分:
gsva_matrix<- gsva(as.matrix(uni_matrix), list,method=’ssgsea’,kcdf=’Gaussian’,abs.ranking=TRUE)
如下图:
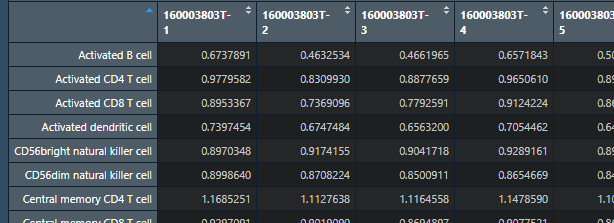
数据已经分析完成,我们后面就将结果可视化,通过富集分数的画热图!
library(pheatmap)
gsva_matrix1<- t(scale(t(gsva_matrix)))#归一化
gsva_matrix1[gsva_matrix1< -2] <- -2
gsva_matrix1[gsva_matrix1>2] <- 2
anti_tumor <- c(‘Activated CD4 T cell’, ‘Activated CD8 T cell’, ‘Central memory CD4 T cell’, ‘Central memory CD8 T cell’, ‘Effector memeory CD4 T cell’, ‘Effector memeory CD8 T cell’, ‘Type 1 T helper cell’, ‘Type 17 T helper cell’, ‘Activated dendritic cell’, ‘CD56bright natural killer cell’, ‘Natural killer cell’, ‘Natural killer T cell’)
pro_tumor <- c(‘Regulatory T cell’, ‘Type 2 T helper cell’, ‘CD56dim natural killer cell’, ‘Immature dendritic cell’, ‘Macrophage’, ‘MDSC’, ‘Neutrophil’, ‘Plasmacytoid dendritic cell’)
anti<- gsub(‘^ ‘,”,rownames(gsva_matrix1))%in%anti_tumor
pro<- gsub(‘^ ‘,”,rownames(gsva_matrix1))%in%pro_tumor
non <- !(anti|pro)##设定三种基因
gsva_matrix1<- rbind(gsva_matrix1[anti,],gsva_matrix1[pro,],gsva_matrix1[non,])#再结合起来,使图分成三段
normalization<-function(x){
return((x-min(x))/(max(x)-min(x)))}#设定normalization函数
nor_gsva_matrix1 <- normalization(gsva_matrix1)
annotation_col = data.frame(patient=pheno$num2)#加上病人编号
rownames(annotation_col)<-colnames(uni_matrix)#使编号能互相对应
bk = unique(c(seq(0,1, length=100)))#设定热图参数
pheatmap(nor_gsva_matrix1,
show_colnames = F,
cluster_rows = F,cluster_cols = F,
annotation_col = annotation_col,
breaks=bk,cellwidth=5,cellheight=5,
fontsize=5,gaps_row = c(12,20),
filename = ‘ssgsea.pdf’,width = 8)#画热图
save(gsva_matrix,gsva_matrix1,pheno,file = ‘score.Rdata’)#不要忘记将结果保存
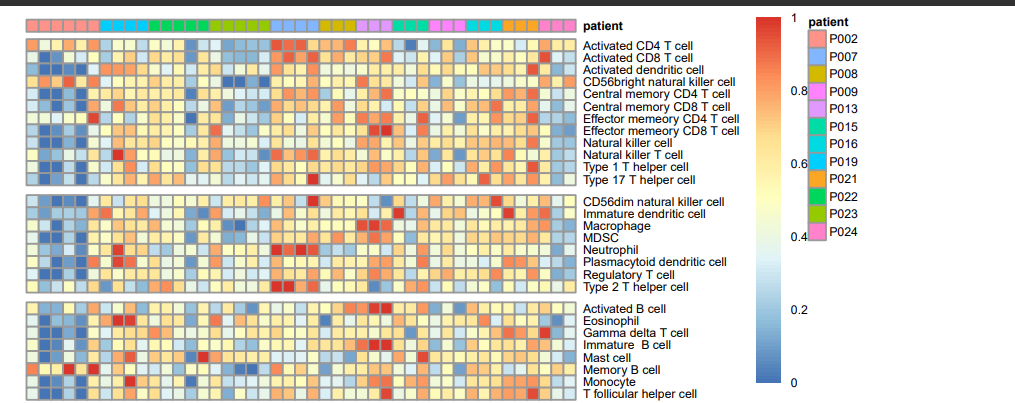 最终我们得到如上的热图绘制
最终我们得到如上的热图绘制
当然,对于免疫细胞的程度分析肯定少不了相关性分析图,我们下面通过ggplot2去绘制一下:
rm(list=ls())
anti_tumor <- c(‘Activated CD4 T cell’, ‘Activated CD8 T cell’, ‘Central memory CD4 T cell’, ‘Central memory CD8 T cell’, ‘Effector memeory CD4 T cell’, ‘Effector memeory CD8 T cell’, ‘Type 1 T helper cell’, ‘Type 17 T helper cell’, ‘Activated dendritic cell’, ‘CD56bright natural killer cell’, ‘Natural killer cell’, ‘Natural killer T cell’)
pro_tumor <- c(‘Regulatory T cell’, ‘Type 2 T helper cell’, ‘CD56dim natural killer cell’, ‘Immature dendritic cell’, ‘Macrophage’, ‘MDSC’, ‘Neutrophil’, ‘Plasmacytoid dendritic cell’)
load(‘score.Rdata’)
anti<- as.data.frame(gsva_matrix1[gsub(‘^ ‘,”,rownames(gsva_matrix1))%in%anti_tumor,])
pro<- as.data.frame(gsva_matrix1[gsub(‘^ ‘,”,rownames(gsva_matrix1))%in%pro_tumor,])
anti_n<- apply(anti,2,sum)
pro_n<- apply(pro,2,sum)
patient <- pheno$num2[match(colnames(gsva_matrix1),pheno$num1)]
library(ggplot2)
data <- data.frame(anti=anti_n,pro=pro_n,patient=patient)
anti_pro<- cor.test(anti_n,pro_n,method=’pearson’)
gg<- ggplot(data,aes(x = anti, y = pro),color=patient) +
xlim(-20,15)+ylim(-15,10)+
labs(x=”Anti-tumor immunity”, y=”Pro-tumor suppression”) +
geom_point(aes(color=patient),size=3)+geom_smooth(method=’lm’)+
annotate(“text”, x = -5, y =7.5,label=paste0(‘R=’,round(anti_pro$estimate,4),’\n’,’p<0.001′))
ggsave(gg,filename = ‘cor.pdf’, height = 6, width = 8)
绘制图的方式原理和上面差不多,
看一下结果把
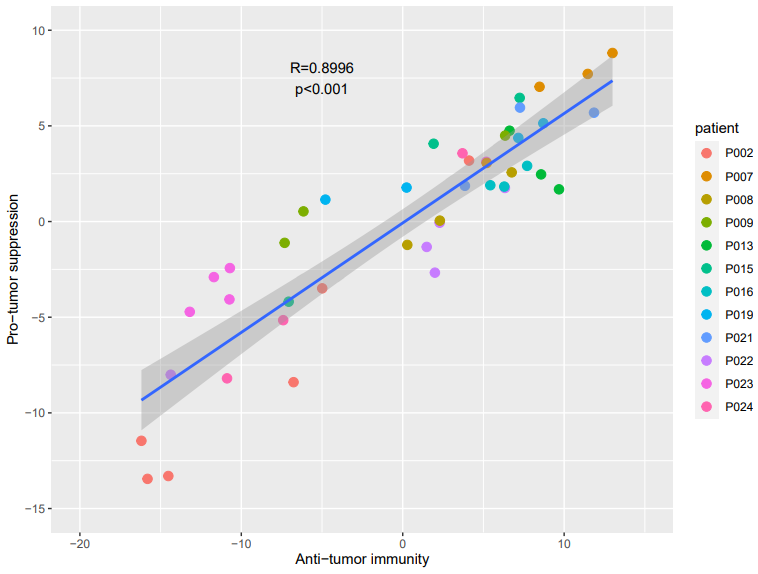
这样一来我们就大功告成了,小伙伴有没有心动呢,快去试试吧
不要忘记多多理解代码的意义这样才能绘制出自己想要的图片!