在生物信息学中,数据处理是非常重要的一环。为了方便进行数据操作和处理,R语言提供了许多强大的包,其中之一就是dplyr包。dplyr包是一个功能强大且易于使用的数据操作包,它提供了一组简洁、一致且易于记忆的函数,用于对数据进行筛选、排序、整理和汇总等操作。下面小果将通过一个实例来介绍如何使用dplyr包进行数据处理,感兴趣的话就和小果一起看下去吧!
了解dplyr
在生物信息学中,dplyr包常常用于对基因表达数据、单细胞转录组数据等进行数据处理和分析。以下是dplyr包在生物信息学中的一些常见应用:
- 数据过滤:使用dplyr的filter()函数可以根据特定条件筛选出感兴趣的基因或样本。例如,可以筛选出差异显著的基因来进行后续分析。
- 数据选择和重命名:使用dplyr的select()函数可以选择感兴趣的列,比如选择特定基因的表达水平。同时,还可以使用rename()函数对列进行重命名,方便后续分析和可视化。
- 数据整理:使用dplyr的mutate()函数可以创建新列,例如计算基因的表达比例或加入其他注释信息。使用arrange()函数可以对数据按特定列进行排序,例如按照基因表达水平排序。
- 数据聚合和汇总:使用dplyr的group_by()函数可以将数据按照特定变量进行分组,然后使用summarize()函数计算每个组的摘要统计量,如平均值、中位数等。这在分析不同条件下的基因表达差异时非常有用。
- 数据合并和连接:使用dplyr的bind_rows()和bind_cols()函数可以将不同样本或实验的数据合并成一个大的数据框,方便后续分析。此外,使用left_join()、inner_join()等函数可以根据共享的列将多个数据框连接起来。
这只是dplyr包在生物信息学中的一些常见应用,实际上,dplyr还提供了更多强大而灵活的功能,可以帮助研究人员进行高效的数据处理和分析,从而更好地理解和解释生物学数据。
dplyr处理数据
加载R包与数据准备
首先,我们需要加载dplyr包,这两个包包含了许多用于数据处理的函数。
library(dplyr)
接下来,我们可以使用dplyr包自带的starwars数据集来演示dplyr包的使用,同学们可以根据自己的学习情况替换成自己感兴趣的数据哦!
我们可以先来查看一下starwars数据集中的数据:
starwars
查看starwars数据集的类型,可以使用以下代码:
class(starwars)
查看starwars数据集的字段名:
colnames(starwars)
查看starwars数据集的大小,即行数和列数:
dim(starwars)

在生物信息学中,我们通常需要对数据进行筛选、排序和整理等操作。下面将介绍一些dplyr包中常用的函数。
筛选数据:filter()
筛选数据是根据某些条件选择特定行或列的过程。在dplyr包中,可以使用filter()函数来筛选数据。以下是一些示例:
# 筛选出height为150的行
filter(starwars, height == 150)
# 筛选出sex为female的行
filter(starwars, sex == ‘female’)
# 筛选出skin_color为light并且height等于150的行
filter(starwars, skin_color == ‘light’ & height == 150)
# 筛选出skin_color为light或者height等于150的行
filter(starwars, skin_color == ‘light’ | height == 150)

排序数据:arrange()
排序数据是根据某一列的值对数据进行升序或降序排列的过程。在dplyr包中,可以使用arrange()函数来排序数据。以下是一些示例:
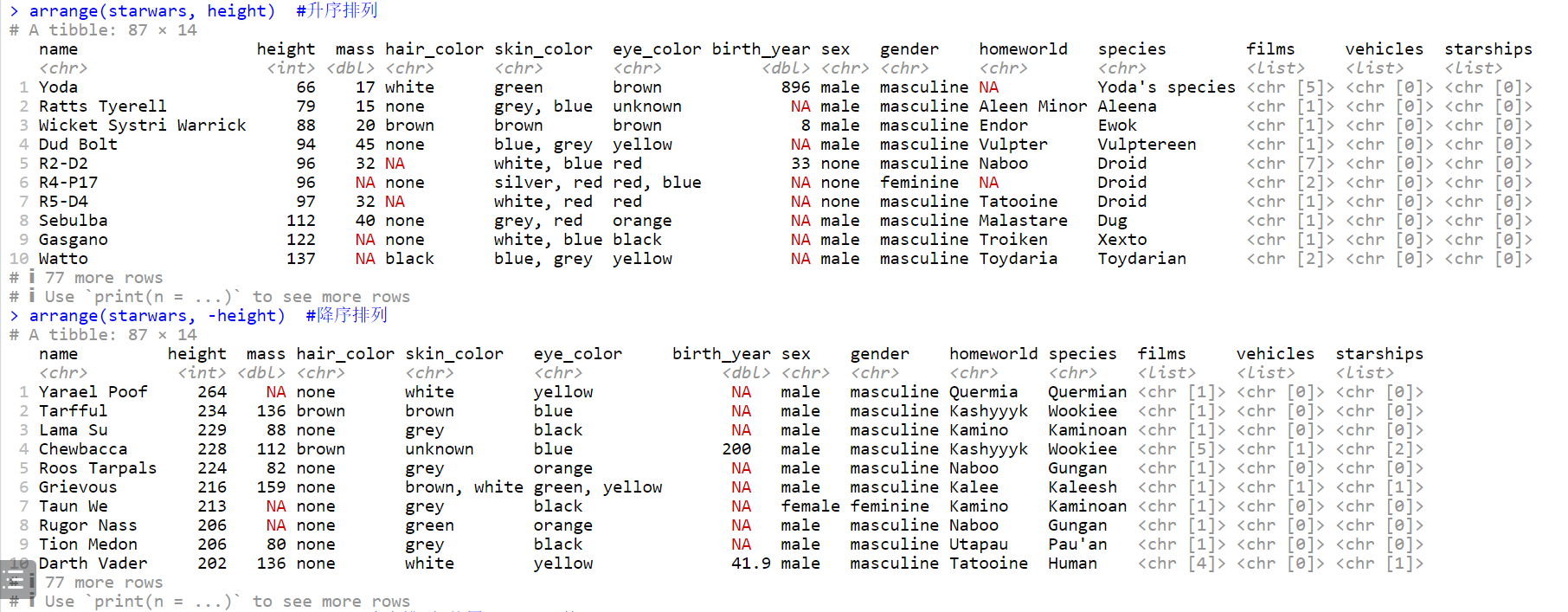
# 升序排列
arrange(starwars, height)
# 降序排列
arrange(starwars, -height)
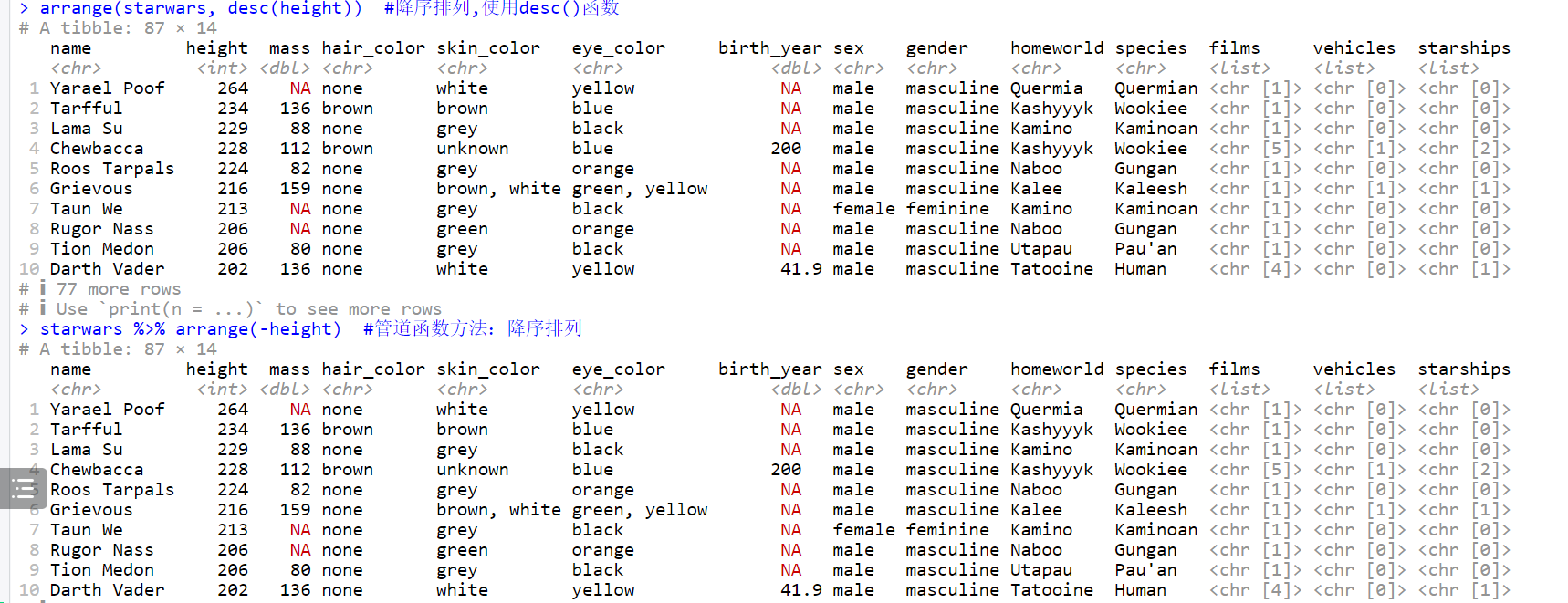
# 降序排列,使用desc()函数
arrange(starwars, desc(height))
整理数据:mutate()
在生物信息学中,有时我们需要添加新的列或修改现有列来进行进一步分析。在dplyr包中,可以使用mutate()函数来实现这个功能。以下是一些示例:
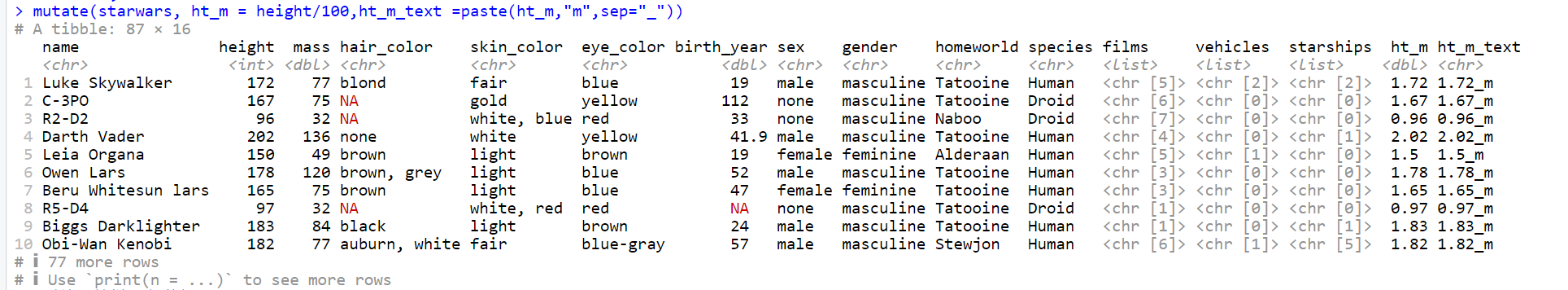
# 添加两列:ht_m将身高数据除以100,color列连接skin_color和eye_color两列
mutate(starwars, ht_m = height/100, color = paste(skin_color, eye_color, sep = “_”))
# 计算新列ht_m和color,并返回仅包含新列的对象
transmute(starwars, ht_m = height/100, color = paste(skin_color, eye_color, sep = “_”))
汇总数据:summarise()
在生物信息学中,我们经常需要计算数据的统计量,如均值、标准差、最大值等。在dplyr包中,可以使用summarise()函数来实现这个功能。以下是一些示例:
# 返回数据中height的均值
summarise(na.omit(starwars), mean(height))
# 返回数据中height的标准差
summarise(na.omit(starwars), sd(height))
# 返回数据中height的最大值和最小值
summarise(na.omit(starwars), max(height), min(height))
# 返回starwars数据框的行数
summarise(starwars, n())
# 返回sex去重后的个数
summarise(starwars, n_distinct(sex))
# 返回height的第一个值
summarise(starwars, first(height))
# 返回height的最后一个值
summarise(starwars, last(height))

以上就是使用dplyr包进行数据处理的一些常用函数和示例。通过学习和掌握这些函数,你可以更加方便地对生物信息学中的数据进行清洗、整理和分析。怎么样,是不是很简单!希望这个教程对你理解dplyr包的使用和生物信息学的应用有所帮助,大家赶快实操起来吧!!