在生物学领域,深入研究DNA、RNA和蛋白质序列是揭示生命机制和演化过程的关键一环。在这个令人着迷的领域中,科学家们不断追求更高效、更准确的方法来解读序列中隐藏的信息,以便了解它们的功能、结构和相互作用。为了满足这一需求,一系列工具和技术应运而生,其中在R语言生态系统中的seqLogo包就扮演着极为重要的角色。seqLogo的出现,为研究人员提供了一种直观、富有信息的方式,用于展示序列的保守性和变异性模式。
seqLogo是一款基于R语言的包,其主要功能在于创造性地构建序列标识图,从而以视觉化的形式将序列在不同位置上的特征显现出来。这些特征主要体现在序列的保守性和变异性,即在不同位置上,序列中的碱基或氨基酸是否具有一定的一致性,或者表现出较高的多样性。这种图形化的呈现方式不仅富有美感,更重要的是,它能够帮助研究人员轻松发现一组相关序列中的重要模式和趋势,进而加深对DNA、RNA和蛋白质序列生物学功能及相互作用的理解。
在seqLogo的背后,蕴藏着信息论的核心概念。信息论是一门研究信息量和信息传递的学科,将碱基或氨基酸视为信息的基本单元,考察它们在特定位置上出现的概率分布。seqLogo巧妙地将信息论引入序列分析中,通过计算每个位置上符号的频率和相对频率,揭示序列中的模式。这一过程中,每个位置上的符号频率被转换成信息熵,即信息的不确定程度。信息熵低的位置表明序列在此处相对保守,而高的信息熵则意味着多样性较大,变异性较高。
seqLogo的工作流程侧重于分析序列中每个位置的符号频率和信息熵。对于一组相关序列,seqLogo将它们统一转换成一个序列集合,并运用信息熵的概念呈现不同位置上的序列模式。在生成的图表中,每个位置上的符号将被分配一个高度,该高度取决于位置上的信息熵和相对频率。这种设计使得图表能够将保守性高的位置展示为较高的符号,而较不保守的位置则表现为较短的符号。
使用seqLogo包的步骤相对简单。首先,你需要在R中安装并加载该包,然后准备相关序列数据。seqLogo支持DNA、RNA和蛋白质序列,你可以根据需要选取相应的数据集。接着,将这些序列转换成适用于seqLogo的格式,例如DNAStringSet对象,之后通过绘图函数即可得到序列标识图。标识图中的每个位置都将以其信息熵和相对频率为基础分配相应的符号,从而形成图形化展示。
要使用seqLogo包,可以在R中使用以下命令进行安装和加载:
> install.packages(“seqLogo”) #安装seqLogo语言包
> library(seqLogo) #加载语言包
通过将信息熵转化为可视化图形,seqLogo以其独特而生动的方式呈现了序列在不同位置上的模式,从而为生物学研究提供了极大的洞察力。在这个引人入胜的图形中,每个序列位置都被赋予了一个视觉高度,这个高度是根据其信息熵和相对频率计算得出的。这种精巧的设计使得图形成为一个视觉图景,准确地传达了序列中的模式信息,使人们能够一目了然地识别出保守模式和变异模式,为进一步的生物学解释和研究提供了有力的工具。
在生成的图表中,较为保守的位置将呈现出更高的符号,这是因为这些位置上的符号具有较低的信息熵。保守性高的位置意味着在这些位置上,序列中的符号倾向于相对一致地出现,形成了稳定的序列模式。这种模式往往与生物学功能密切相关,例如在蛋白质结构中的保守位点,或者在基因的启动子区域中的保守DNA序列。图表中高度的增加使得这些保守模式能够引人注目,帮助研究人员更准确地识别出具有重要生物学功能的序列片段。
与此相反,在图表中,较不保守的位置将表现为更短的符号,因为这些位置上的符号具有较高的信息熵。这表示在这些位置上,序列的变异性较大,符号的出现相对不稳定。这些变异模式可能与不同物种、亚型或变异有关,也可能代表了序列中的无序区域。图表中的这种短符号可以快速吸引注意力,帮助研究人员确定序列中的这些变异模式,进而探索其在生物学上的潜在意义。
示例:
#首先,确保你已经安装了seqLogo包
> install.packages(“seqLogo”)
#两个DNA序列作为示例数据
> sequence1 <- “ATGCTAAGCGTA”
> sequence2 <- “ATGCAAAGCGTA”
# 导入seqLogo包
> library(seqLogo)
# 创建序列集合
> sequences <- c(sequence1, sequence2)
# 将序列集合转化为DNAStringSet对象
> seq_set <- DNAStringSet(sequences)
# 创建序列标识图
> logo <- seqLogo(seq_set)
# 显示序列标识图
> print(logo)
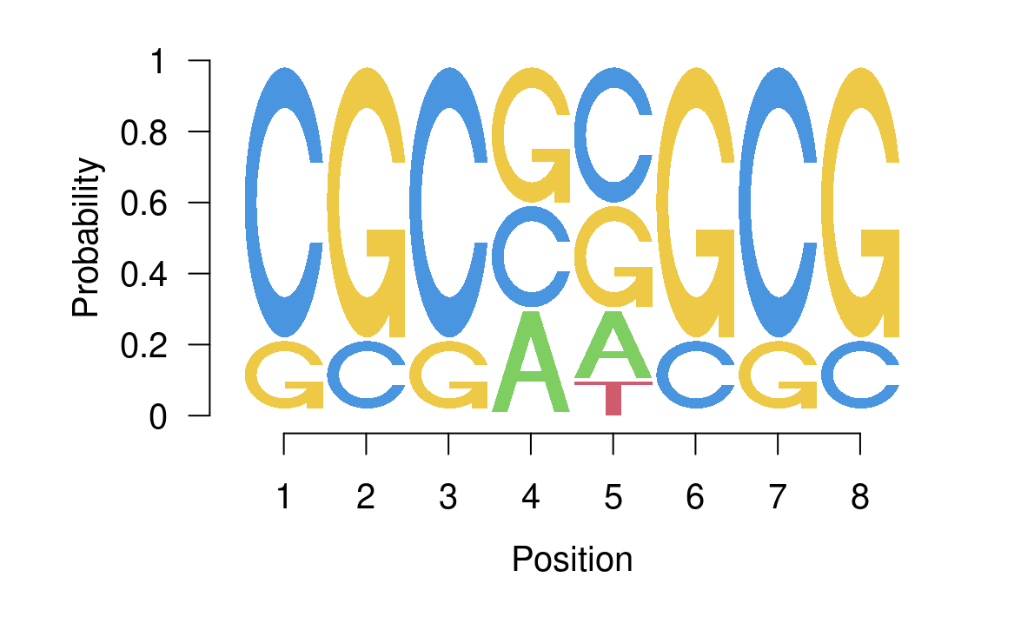
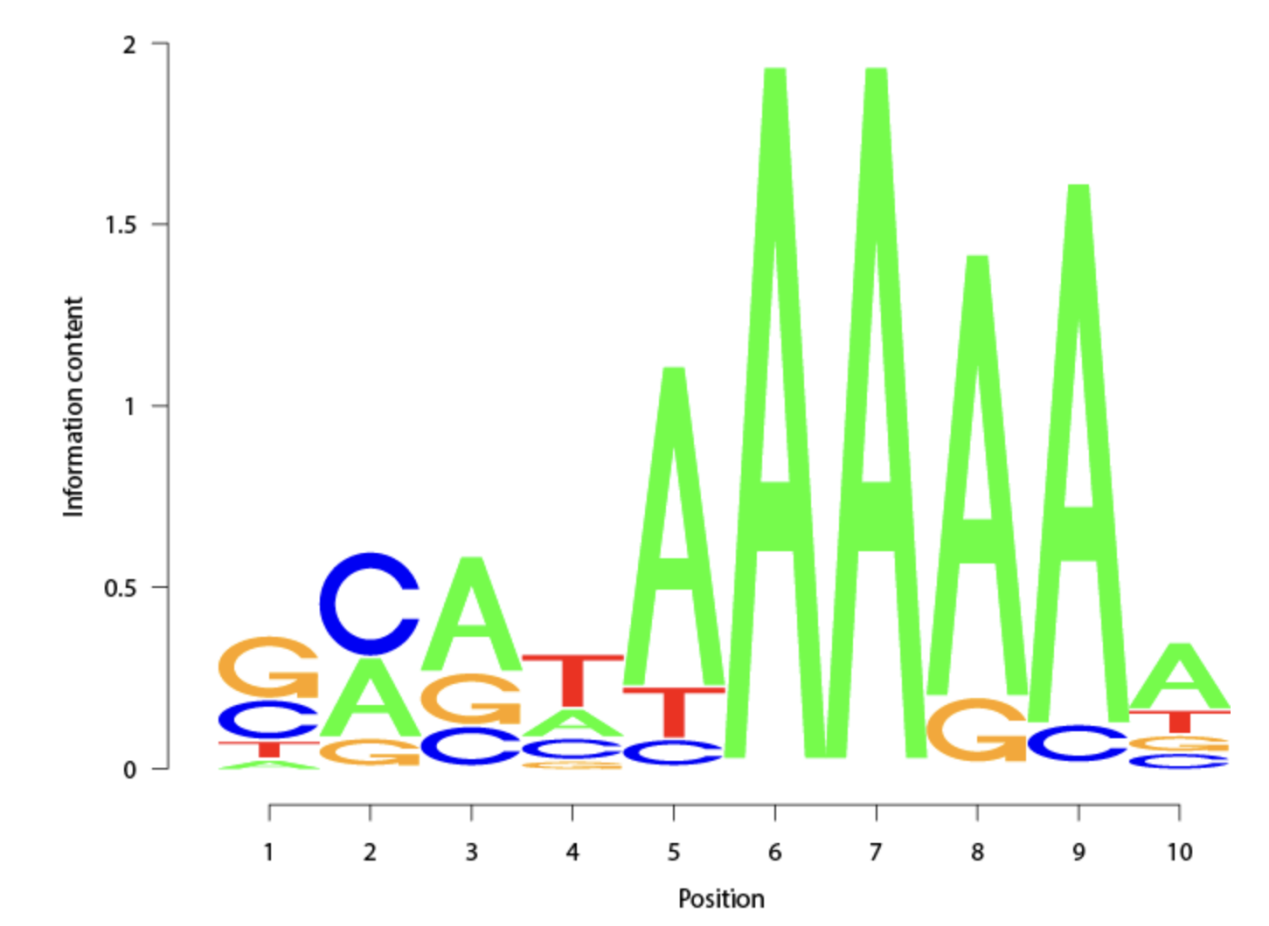
上述代码将生成一个包含序列标识图的绘图窗口。图表中每个位置上的碱基都根据其频率和信息熵来表示。在图表中,较保守的位置会有更高的字符,而较不保守的位置会有较短的字符。通过这个图表,你可以直观地看到这两个序列中的共同模式和差异。
此外,你还可以根据需要进行自定义,例如修改颜色、字体和标签等,以便更好地适应你的分析需求。
这个示例展示了如何使用seqLogo包来可视化DNA序列的模式。在实际应用中,你可以根据需要加载更多序列,并进一步分析它们之间的共同特征和变异。通过这种方式,你可以更好地理解生物序列中的保守性和变异性,为生物学研究提供更深入的见解。
标识图的解读能力在于图形的高度分配。较为保守的位置将呈现出更高的符号,象征着在这些位置上的序列模式相对稳定。这些保守模式通常与生物学功能密切相关,例如在蛋白质结构中的关键位点,或基因启动子中的保守DNA序列。这些位置在图中的显著高度有助于直观地辨识这些重要模式。
与此同时,较不保守的位置将显示为较短的符号,这源自这些位置上的信息熵较高。这表示在这些位置上,序列呈现出较大的变异性,符号的出现更加不稳定。这些变异模式可能与不同物种、亚型或变异相关,也可能对应序列中的无序区域。图表中的短符号引人注目,有助于揭示这些变异模式,进而深入探索其在生物学上的潜在意义。
通过将复杂的序列信息转换成直观的标识图,seqLogo帮助生物学家更深入地洞察序列的模式和特征。它不仅在解释DNA、RNA和蛋白质序列中的生物学功能和相互作用方面发挥着重要作用,还为整个生命科学领域的研究提供了宝贵的见解。因此,seqLogo无疑是生物信息学领域的一颗明星,引领着研究者们走向更深远的序列解读之路。
seqLogo不仅是一个有力的工具,也是一个概念性的桥梁,将信息论与生物学相结合,使研究人员能够更好地理解序列的特征。通过这种方式,研究人员可以更加深入地挖掘DNA、RNA和蛋白质序列中的生物学信息,为生命科学领域的研究提供更精准和全面的视角。
seqLogo通过以人类易于理解的方式呈现序列模式,极大地促进了生物学家们对序列保守性和变异性的认识。这种视觉化方法不仅使得复杂的序列数据变得更加直观和可解释,还为生物学研究提供了深刻的洞察,有助于揭示序列的生物学功能、相互作用和进化特征。
以上就是对R语言包seqLogo的简单介绍啦,seqLogo为生物学家们提供了一个强大的工具,帮助他们分析和理解生物序列的模式和特征。通过信息论的原理,seqLogo能够将复杂的序列信息转化为直观的图形,使研究人员能够更好地解释序列的保守性和变异性。无论是研究DNA、RNA还是蛋白质序列,seqLogo都为生物学领域的研究带来了更深入的洞察力。
总的来说,seqLogo作为R语言中的重要工具,以其独特的信息论基础和图形化呈现方式,通过将复杂的序列信息转换成直观的标识图,seqLogo帮助生物学家更深入地洞察序列的模式和特征。它不仅在解释DNA、RNA和蛋白质序列中的生物学功能和相互作用方面发挥着重要作用,还为整个生命科学领域的研究提供了宝贵的见解。因此,seqLogo无疑是生物信息学领域的一颗明星,引领着研究者们走向更深远的序列解读之路。
小伙伴们,今天有没有学到新知识呢,想要继续了解R语言内容可以持续关注小果哦~~或者也可以关注我们的官网也会持续更新的哦~ http://www.biocloudservice.com/home.html
References: