在上一篇分享中,小果给大家详细地分享了分子对接的相关知识以及简单的分子对接的教程,那么这一次小果就来给大家更加详细地介绍一下dock ligands模块中各个功能的作用啦。废话不多说,我们直接开始吧!
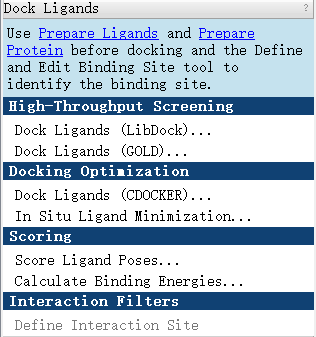
“LibDock”、”GOLD”和”CDOCKER”
首先,前三个功能”LibDock”、”GOLD”和”CDOCKER”是三种分子对接软件,它们的简要介绍如下:
- “LibDock”是一种常用的基于晶格匹配和特征评分的对接方法,它使用小分子库进行高通量筛选,通过判断分子与蛋白质之间的亲和力来预测结合能力和亲合性。
- “GOLD”(Genetic Optimization for Ligand Docking)是一种基于遗传算法的对接程序,它通过在分子内搜索合适的位姿和构象来寻找最佳的配体-蛋白质结合方式。”GOLD”使用分子力学力场和评分函数进行评估,并利用遗传算法进行优化。
- “CDOCKER”(Chemical Docking)是一种基于药物设计的自动化对接软件,它使用分子对接算法来预测小分子与蛋白质之间的相互作用。”CDOCKER”使用的是一种基于能量和相互作用的评估指标,以确定最佳的配体-蛋白质结合模式。
这里小果再对三种分子对接软件的适用情况、特点和优劣进行简要说明:
LibDock:
- 适用情况:适用于高通量筛选,从大型小分子库中快速筛选出可能的结合配体。
- 特点:基于晶格匹配和特征评分,速度较快,适用于大规模筛选。
- 优劣:
- 优点:计算速度快,适用于高通量筛选;易于使用和实施。
- 缺点:较简化的评分函数,可能无法捕捉到复杂的相互作用;结果可能有一定误差。
GOLD:
- 适用情况:适用于预测精确的配体-蛋白质结合位姿,并优化配体的构象。
- 特点:基于遗传算法,能够搜索多种配体位姿和构象,并通过评分函数进行优化。
- 优劣:
- 优点:能够找到更准确的配体结合位姿和构象;适用于结合模式较复杂的系统。
- 缺点:计算耗时较长;需要更多的参数和设置,较复杂。
CDOCKER:
- 适用情况:适用于药物设计和药物优化过程中的自动化对接。
- 特点:基于能量和相互作用评估配体-蛋白质结合,支持分子力学模拟。
- 优劣:
- 优点:适用于药物设计过程,能够考虑能量和相互作用;能够进行分子力学模拟。
- 缺点:需要较长的计算时间;结果可能受到力场参数的影响。
说到这里,有的小伙伴可能会产生疑问,既然它们都是分子对接软件,那么为什么libdock和gold属于high-throughput screening分栏,而CDOCKER属于docking optimization分栏呢?要说清楚这个,小果就需要带着大家查阅官方文档,从这两个分栏说起:
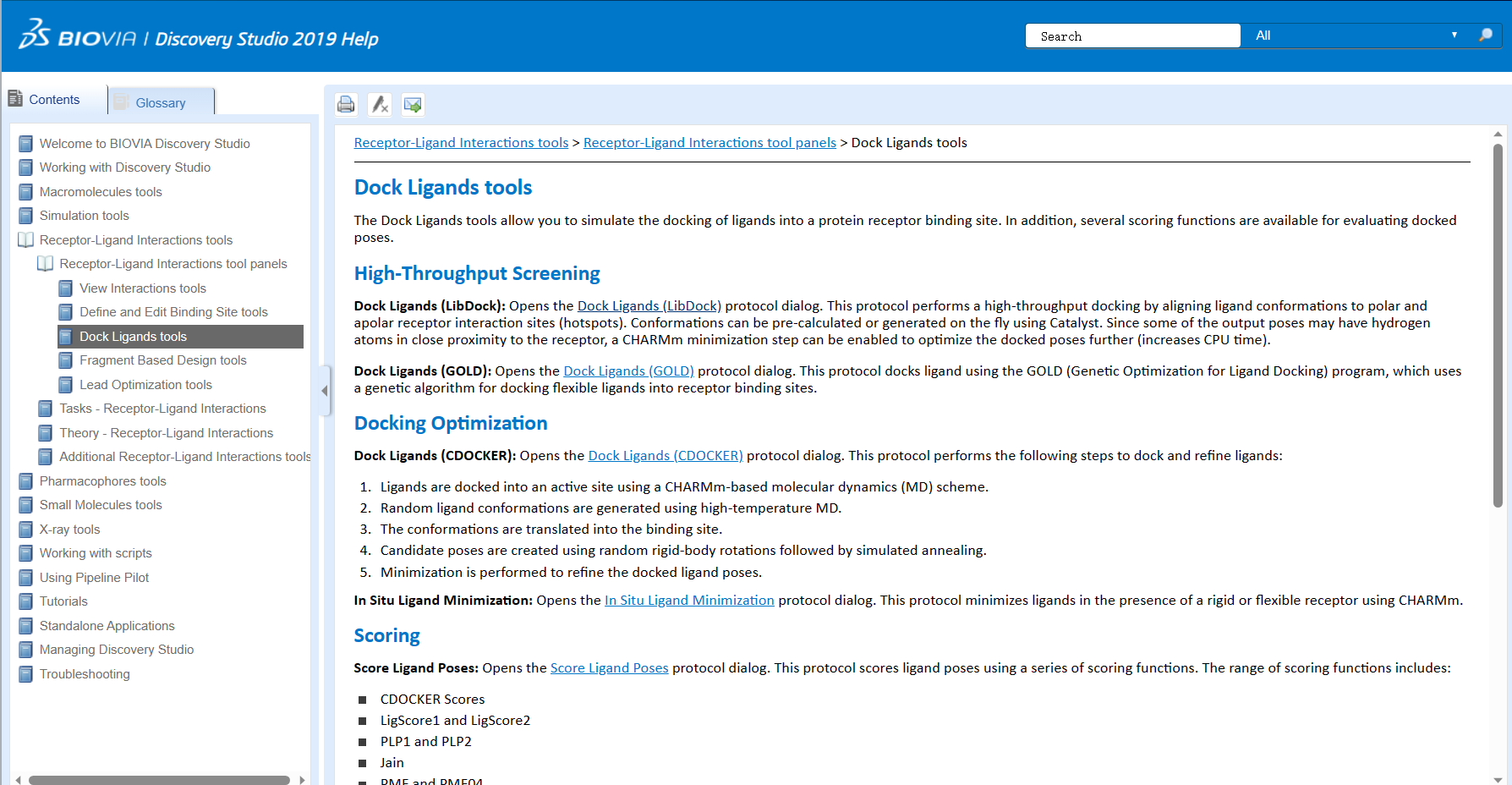
High-throughput screening与Docking optimization
High-throughput screening(高通量筛选)是一种药物发现和化合物筛选的技术和方法。它通过自动化和并行化处理大规模化合物库,快速测试和评估潜在的药物分子或化合物集合与特定生物目标之间的相互作用。
传统上,在药物发现过程中,研究人员需要手工逐个测试每个化合物与特定生物靶点的相互作用。这种方法非常耗时且费力,并且限制了药物筛选的规模和效率。
而高通量筛选则通过使用自动化设备和机器学习算法,能够以非常高的速度和效率对数千到数百万个化合物进行筛选。该技术可以在短时间内评估大量化合物的活性、选择性和毒理学特性,从而更快地发现具有潜在药物活性的化合物。
高通量筛选通常涉及以下步骤:
- 化合物库准备:构建或选择包含大量化合物的库,这些化合物可能具有药物活性。
- 实验操作:将化合物库中的化合物逐个或以平行方式添加到生物试剂中,如酶、受体、细胞等。
- 高通量检测:使用自动化仪器对每个化合物进行快速测试和评估,以确定其与目标的相互作用或活性。
- 数据分析:使用计算方法和机器学习算法对筛选结果进行处理和分析,识别出具有潜在药物活性的化合物。
高通量筛选技术在药物发现领域中起到了关键作用。它能够加速药物发现过程,提高筛选效率,并使研究人员能够更快地发现新的潜在治疗药物。同时,高通量筛选也为大规模的化合物库提供了全面的评估,有助于挖掘更多的药物候选化合物。
而Docking optimization(对接优化)是一种用于分子对接模拟的计算方法,旨在预测蛋白质和小分子之间的结合方式和亲和力。通过对接优化,可以研究蛋白质和小分子之间的相互作用,从而更好地理解它们之间的结合机制和影响因素。
对接优化通常包括以下步骤:
- 蛋白质和小分子的准备:首先,需要准备蛋白质和小分子的三维结构。蛋白质结构可以通过X射线晶体学、核磁共振等实验方法获取,或者从蛋白质数据库中获取。小分子可以通过化学合成或从化合物库中选择。
- 初始对接:将小分子的结构与蛋白质的结构进行初始对接。初始对接通常使用基于物理和几何约束的算法,将小分子放置在蛋白质结构中的可能位点附近。
- 结合能评估:评估小分子和蛋白质之间的结合能。这通常涉及计算和优化化学键能、静电相互作用、范德华力等相互作用能。
- 优化和采样:通过调整小分子的构象和蛋白质的柔性来优化对接位姿。这可以通过分子动力学模拟、蒙特卡洛方法等进行。
- 结果分析:对经过优化的对接位姿进行评估和分析,选择最有可能的结合位点和位姿。这可能涉及到对键合模式、氢键网络、疏水相互作用等的分析。
对接优化的目标是寻找最佳的蛋白质-小分子结合位姿,并预测它们之间的结合亲和力。这对于药物设计和药物发现非常重要,因为药物的活性通常与其在靶点上的结合方式和亲和力密切相关。
看到这里,小伙伴们虽然应该学会了这两大专栏各自的侧重和特点,但还不能很直观地理解两大专栏的区别在哪里,现在小果就给大家说一说它们的区别:
- High-Throughput Screening(高通量筛选): HTS分栏主要关注快速且高通量地筛选大量化合物,以发现具有潜在药物活性的候选分子。该分栏提供了一系列工具和方法,用于处理和评估化合物库。通过高效的计算和筛选流程,HTS可帮助用户迅速缩小化合物范围,并优先考虑具有较高结合亲和力或药效的化合物。其中常用的工具包括虚拟筛选、化学指纹分析等。
- Docking Optimization(对接优化): Docking Optimization分栏用于进一步优化和改善分子对接的结果。它主要关注如何通过调整配体的构象和位姿来提高配体与蛋白质的结合能和准确性。这一分栏提供了针对分子对接的多种工具和算法,如基于引导式模拟和分子动力学模拟的方法,以及结合了遗传算法的优化方法。用户可以使用这些工具来进行系统性的对接优化,以获得更可靠和准确的分子结合模型。
因此,HTS分栏主要用于快速筛选和初步评估大量化合物,找到具有潜在活性的候选分子;而Docking Optimization分栏则专注于对初始对接结果进行进一步优化,以提高对接精度和可靠性。两个分栏在药物发现过程中扮演不同的角色,帮助用户进行不同层次和目标的工作。
从我们使用者的角度来说,大家可以理解为需要先进行HTS分栏的操作,然后在使用Docking Optimization分栏进行优化,这么说完小伙伴们肯定可以理解透彻并且融汇贯通啦。
回到我们最初的问题,为什么libdock和gold属于high-throughput screening分栏,而CDOCKER属于docking optimization分栏呢?那是因为它们在分子对接过程中的应用和重点略有不同:
“Libdock”是一种基于锁定-解锁机制的算法,在分子对接过程中使用静态能量评分来评估配体和蛋白质结合位点之间的亲和力。它可以在较短的时间内对大量化合物进行高通量筛选,从而帮助筛选出具有潜在药物活性的化合物。
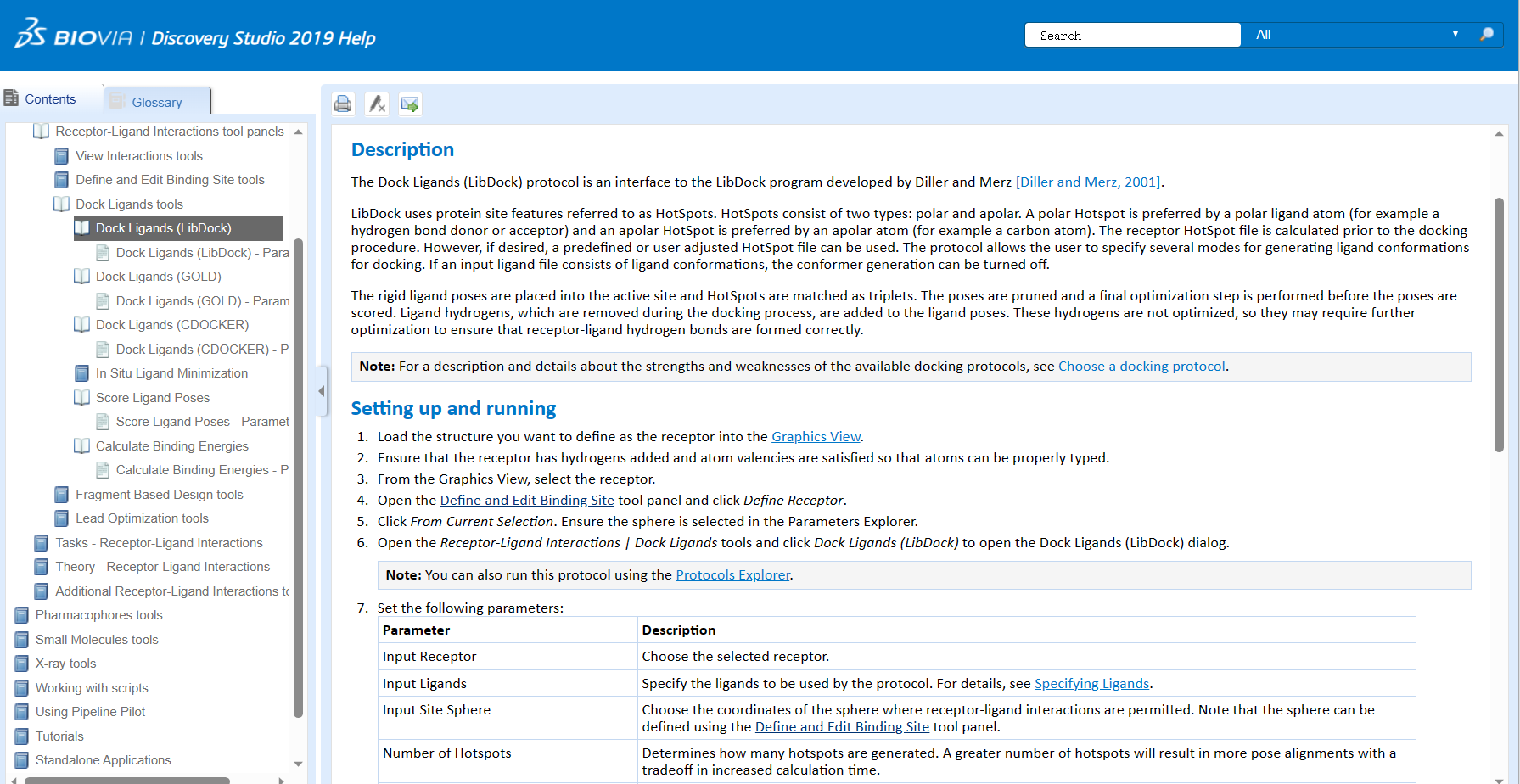
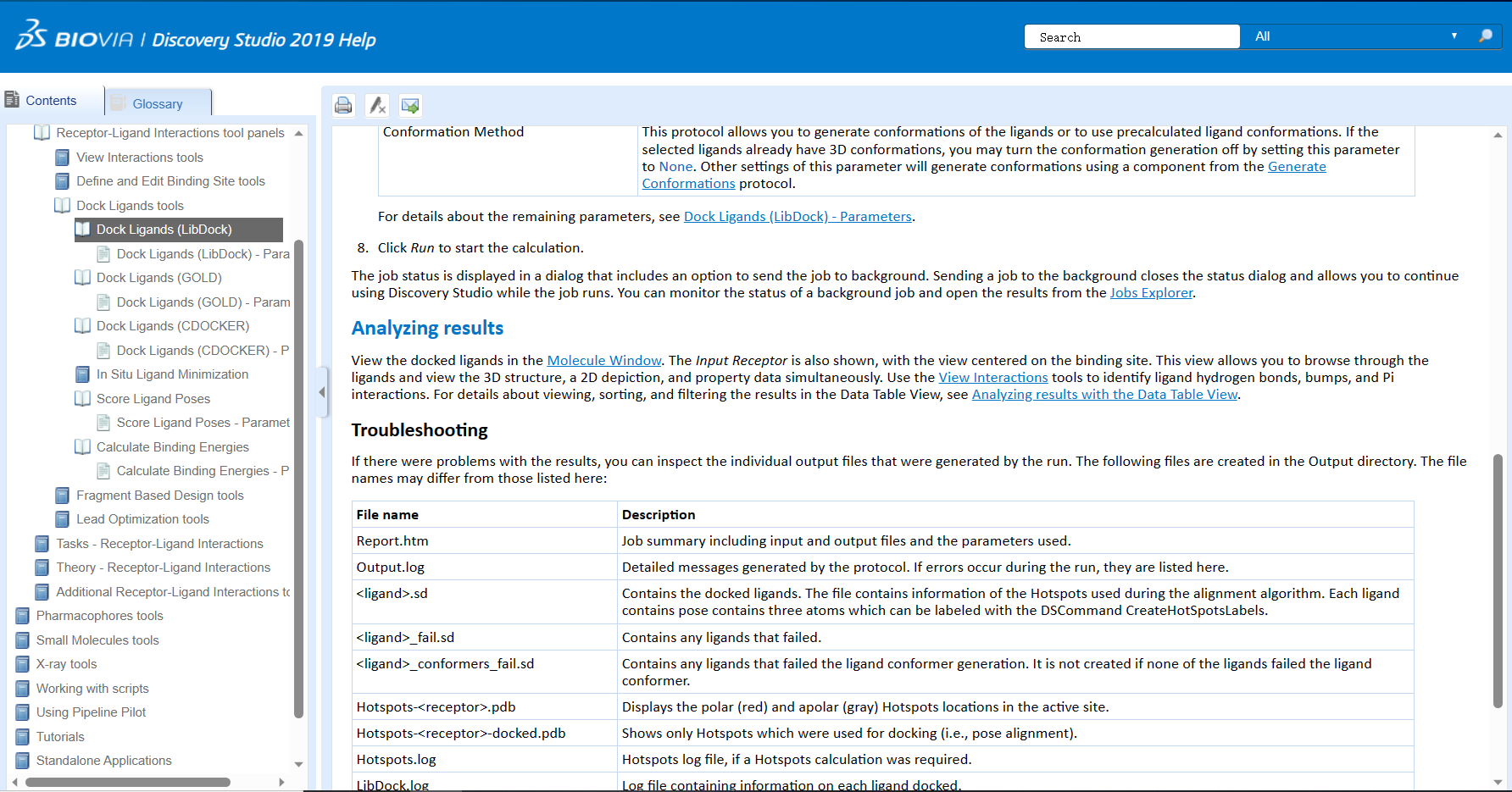
“GOLD”(Genetic Optimization for Ligand Docking)是一种基于遗传算法的分子对接程序,通过优化配体的构象和位姿来寻找最佳的配位模型。它可以处理多样性较高的化合物库,并在对接过程中使用多种评分函数来预测配体与蛋白质的结合能。
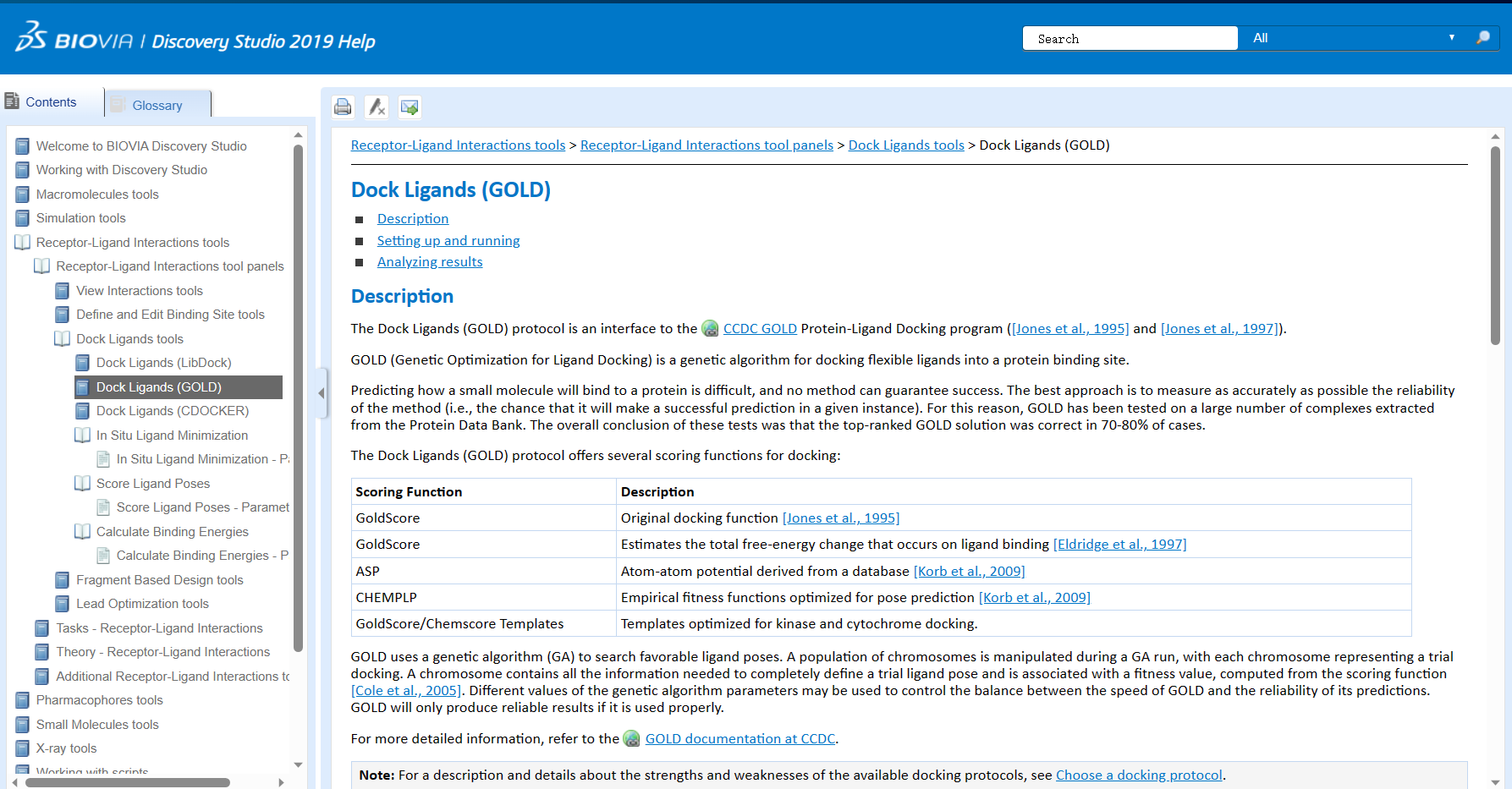
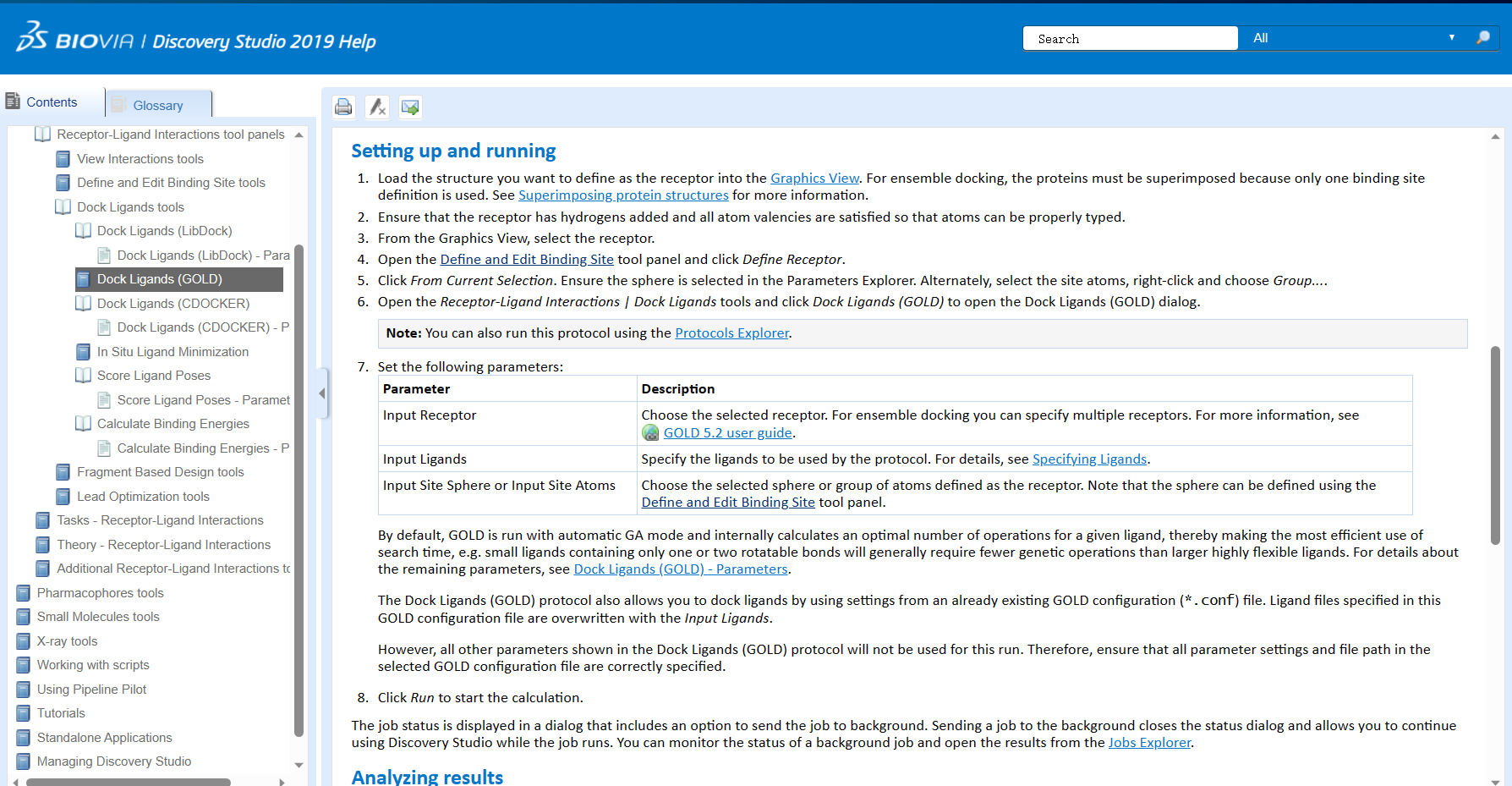
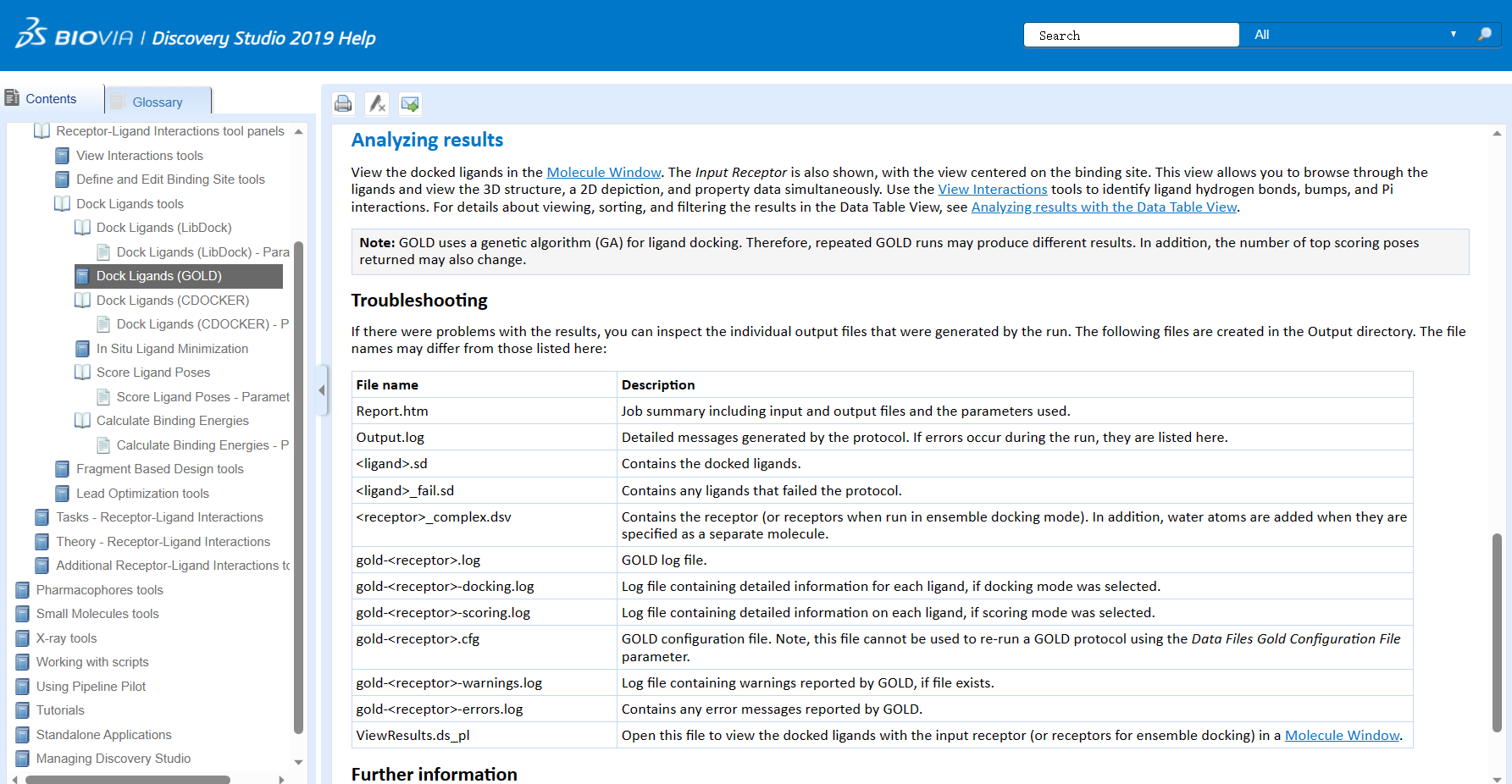
相比之下,”CDOCKER”是一种分子对接优化方法,着重于通过模拟分子动力学的方式对配体进行优化。它主要用于从初始的分子对接结果出发,进一步优化和改善配体的构象和能量,以提高分子对接的准确性。
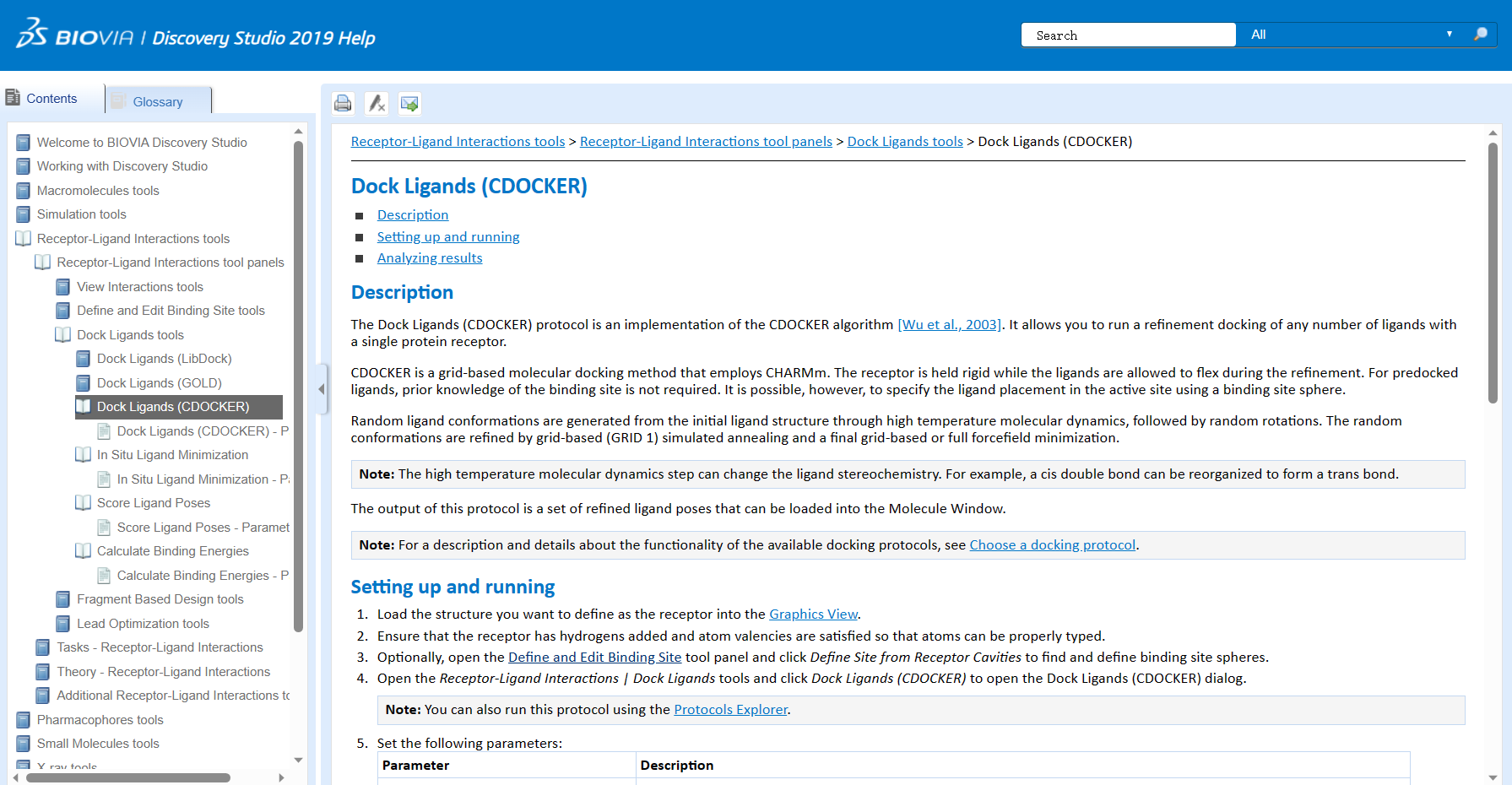
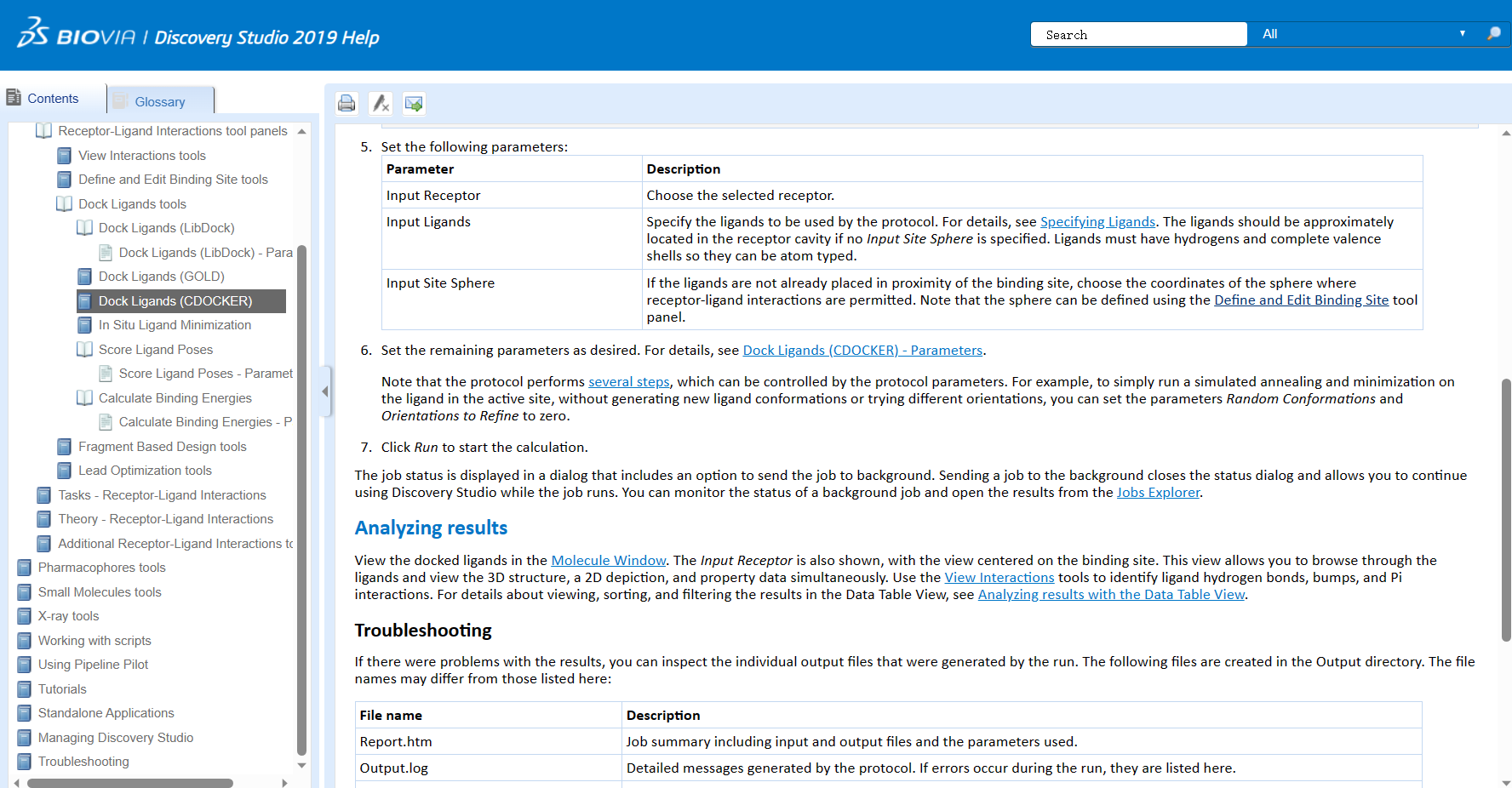
因此,虽然”Libdock”、”GOLD”和”CDOCKER”都与分子对接相关,但它们在应用和重点上有所区别,因此被分为不同的功能分栏。”Libdock”和”GOLD”更适合用于高通量筛选和快速评估候选分子,而”CDOCKER”则更适合用于对接结果的后续优化和调整。
说到这里,大家应该已经明白了他们哥仨之间的区别和联系,现在是不是想认识一下新朋友了呢?那么小果现在就介绍一下Docking optimization下的另一大功能:In situ ligand minimization。
In situ ligand minimization和CDOCKER
我们在分子对接完成后,为了优化配体的构象和能量,也可以使用”In situ ligand minimization”来进行后续处理,它用于对配体进行原位最小化。
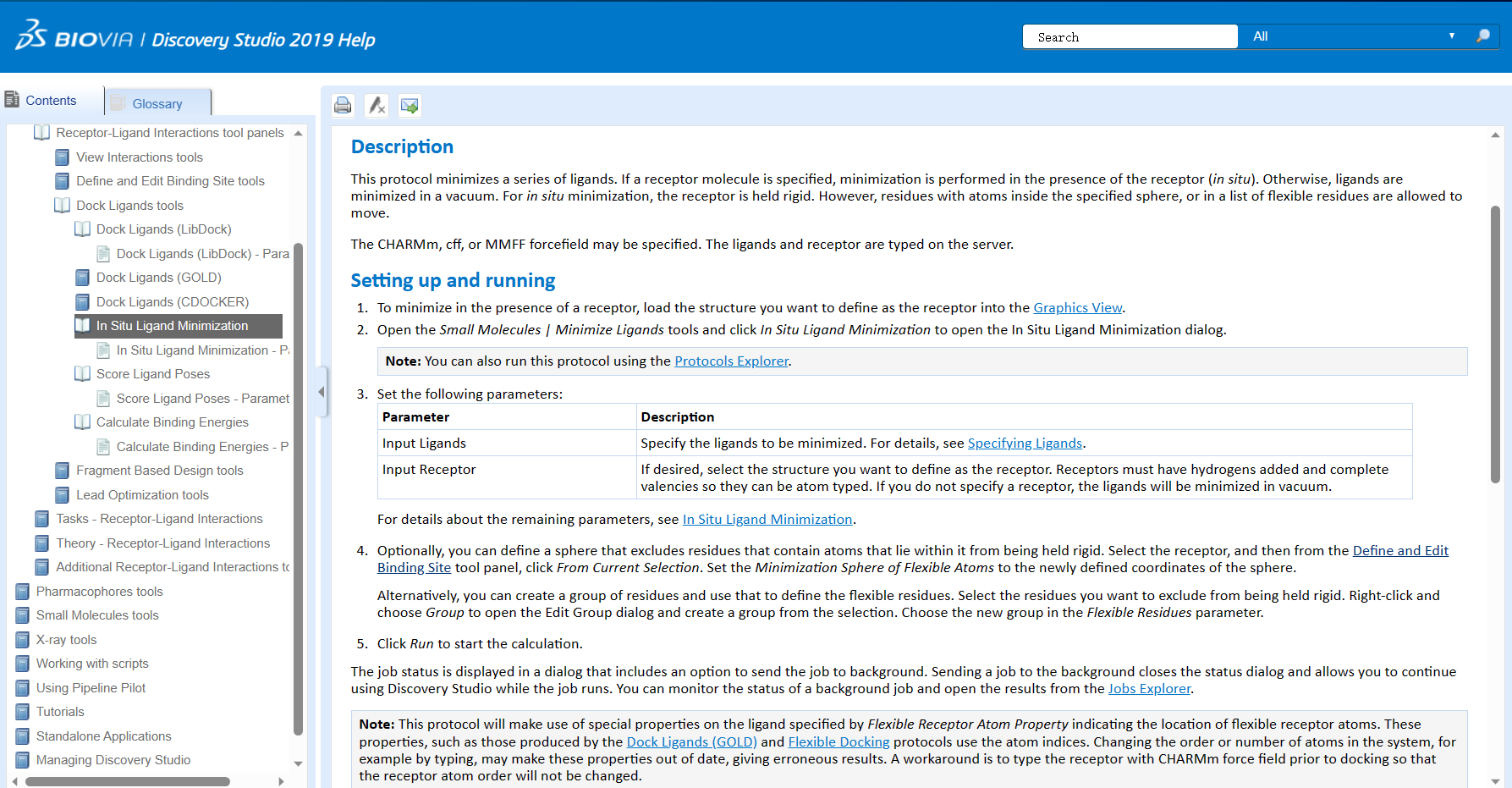
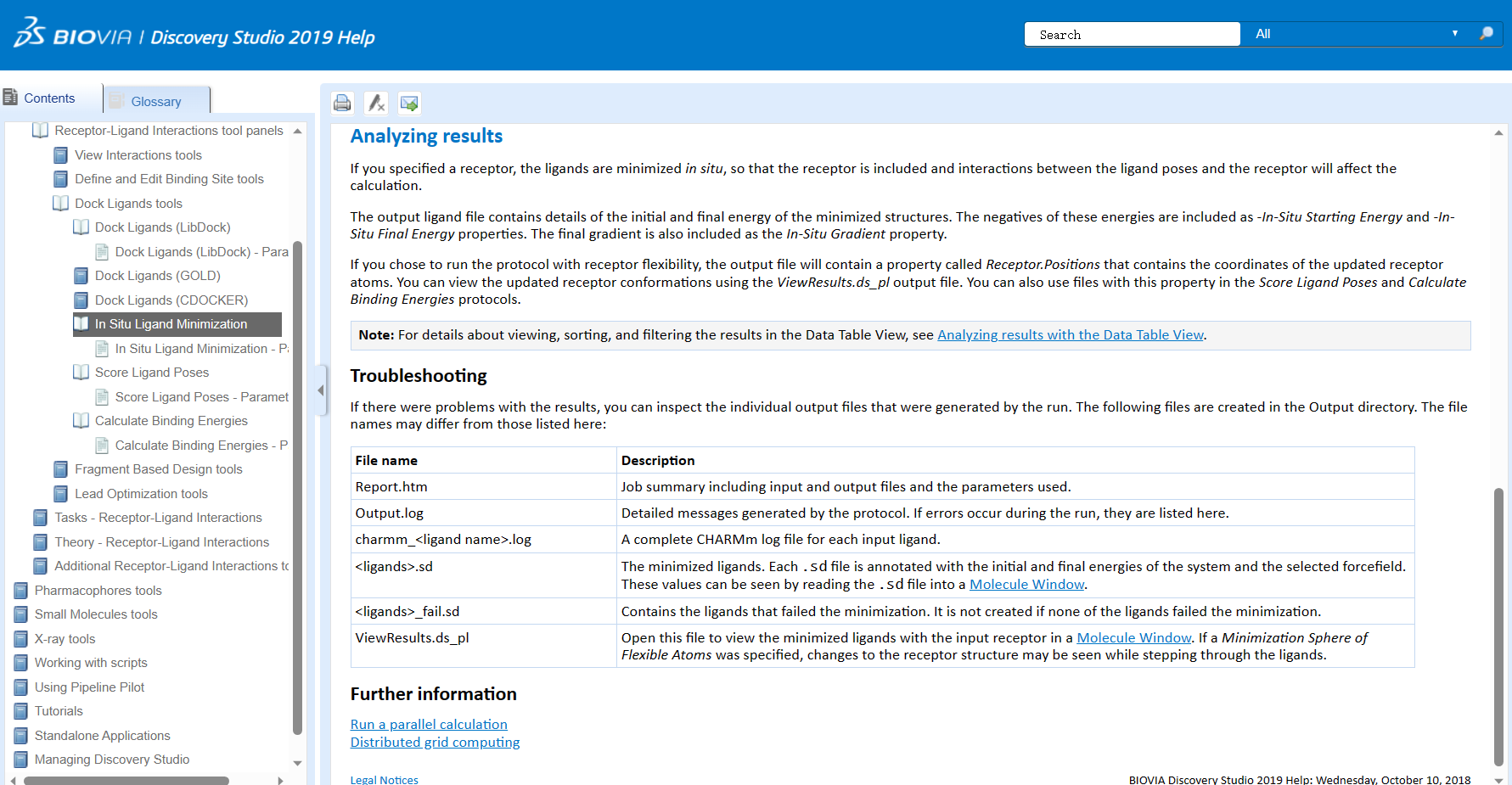
使用”In situ ligand minimization”时,Discovery Studio将对已经对接到蛋白质结构上的配体进行优化。该过程包括以下步骤:
- 能量最小化:通过使用优化算法和力场参数,Discovery Studio会对配体的构象进行调整和优化,以达到能量最低状态。这通常涉及到调整键角、化学键长度、扭转角度等。
- 位点冲突修正:如果配体与蛋白质结构存在位点冲突,即二者之间的原子之间存在空间上的重叠,Discovery Studio会尝试通过调整配体的构象或引入水分子等方式来修复冲突。
- 结果评估:经过”In situ ligand minimization”后,Discovery Studio会提供优化后的配体构象和能量信息。这有助于评估配体的稳定性和结合性能。
使用”In situ ligand minimization”可以优化配体的构象,使其更适合与蛋白质结合,并提高其结合亲和力和稳定性。这有助于进一步的药物设计和分子对接工作。
小果明白大家可能看着这么专业的描述有些头晕眼花,现在小果就把他们的区别详细地写在这里:
- 方法原理:In situ ligand minimization是通过应用能量最小化算法对单个配体进行优化,调整其构象和能量,以改善与蛋白质的结合性能。CDOCKER则使用引导式分子动力学模拟方法,在结合位点中模拟配体的动力学行为,通过搜索位构象并考虑动力学行为来优化配体的位姿和构象。
- 优化策略:In situ ligand minimization主要关注配体的构象和能量的优化,通过能量最小化算法寻找更低能量状态的构象。CDOCKER则综合考虑配体的位构象、能量和动力学行为进行优化,利用分子动力学模拟和遗传算法来搜索配体的更优位构象。
- 考虑因素:In situ ligand minimization主要关注配体的静态性质,如构象和能量,以改善静态结合性能。CDOCKER更关注配体的动态性质,通过模拟动力学行为来优化配体的位构象,考虑配体在结合位点内的灵活性和动态变化。
综上所述,In situ ligand minimization主要通过调整配体的构象和能量进行优化,而CDOCKER则利用分子动力学模拟方法,并综合考虑配体的位构象、能量和动力学行为。两种方法在优化策略和所考虑的因素上有所不同,小伙伴们可根据需求选择合适的方法进行分子对接的优化工作。小果现在就给大家举几个他们适用的例子来更直观地让大家感受一下:
In situ ligand minimization(原位配体最小化)适用于以下情况:
- 初始结构优化:当已有一个初始的配体-蛋白质结合模型时,可以使用原位配体最小化来进一步优化配体的构象和能量,改善结合性能。
- 配体修饰:当需要对已知的配体进行修饰以获得更好的结合性能时,可以使用原位配体最小化来通过调整构象和能量来评估和改善新的配体构象。
- 蛋白质突变分析:在研究蛋白质突变对配体结合的影响时,可以使用原位配体最小化来比较不同突变体的配体结合能力,从而评估突变对结合性能的影响和机制。
而CDOCKER适用于以下情况:
- 高度柔性的配体:当配体具有高度柔性性质,或者在结合过程中可能发生显著的构象变化时,CDOCKER可以更全面地考虑配体的动态性质和灵活性,通过模拟动力学行为来优化配体的位构象。
- 多个配体分子对接:当需要对多个配体分子进行对接优化时,CDOCKER能够利用分子动力学模拟和遗传算法来搜索多个配体的更优位构象,并考虑它们之间的相互作用。
- 结合位点预测:在进行结合位点预测时,CDOCKER可以用于预测配体在潜在结合位点中的位构象,进而评估配体与蛋白质的结合性能。
score ligand poses与calculate binding energies
在分子对接结束后,我们需要对结果进行分析。这里首先就需要用到scoring分栏下面的两个功能:score ligand poses和calculate binding energies。
- Score Ligand Poses: 这个功能用于评估分子在蛋白质激活位点中的不同构象的得分或评分。在分子对接之后,会生成多个可能的配体构象(poses),Score Ligand Poses功能可以使用特定的评分函数对这些构象进行打分。评分函数通常基于配体的几何形状、电荷分布、疏水性以及与蛋白质的相互作用等因素来预测配体与蛋白质之间的结合能。根据分数,可以选择最有可能具有良好结合能的配体构象进行后续分析。
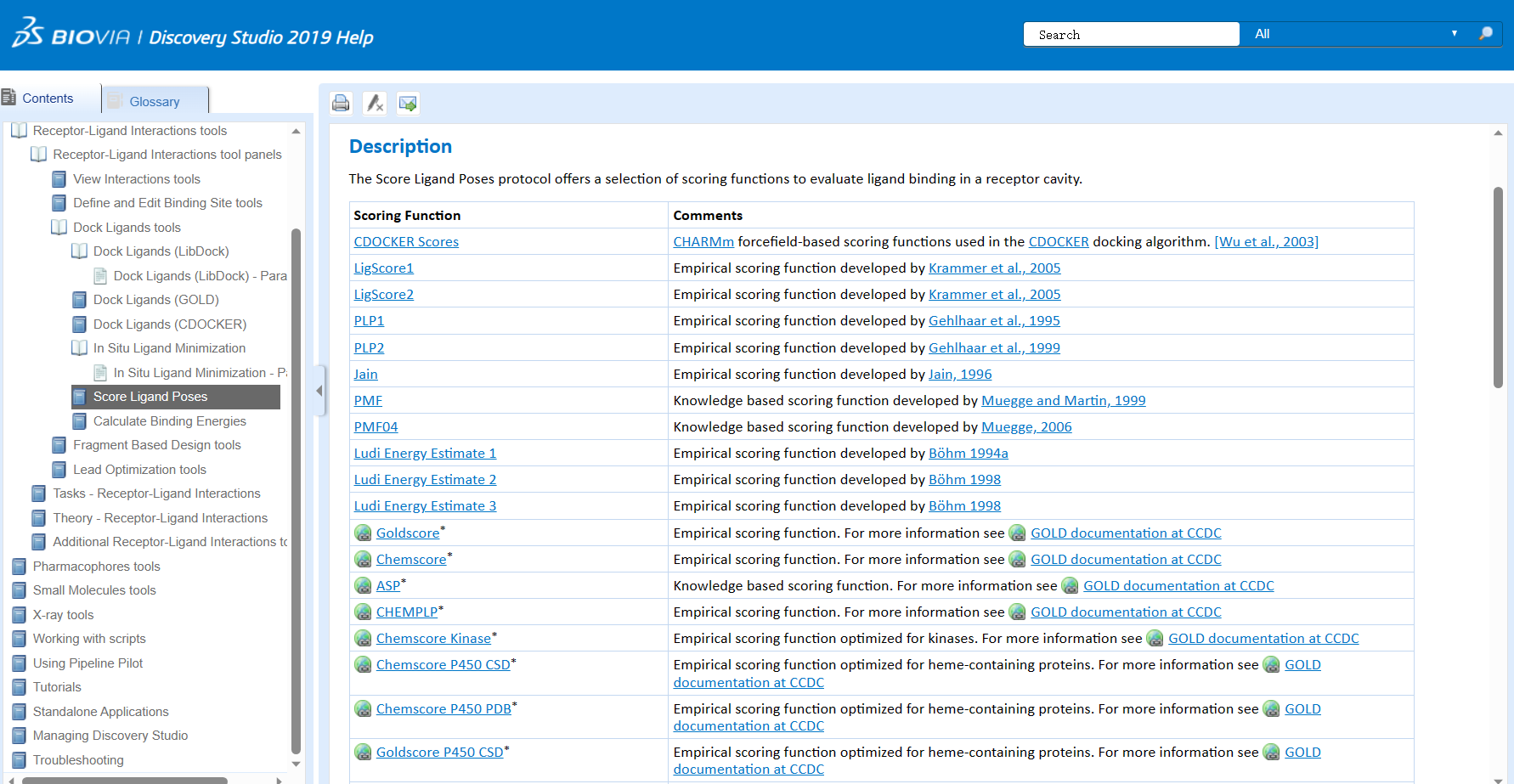

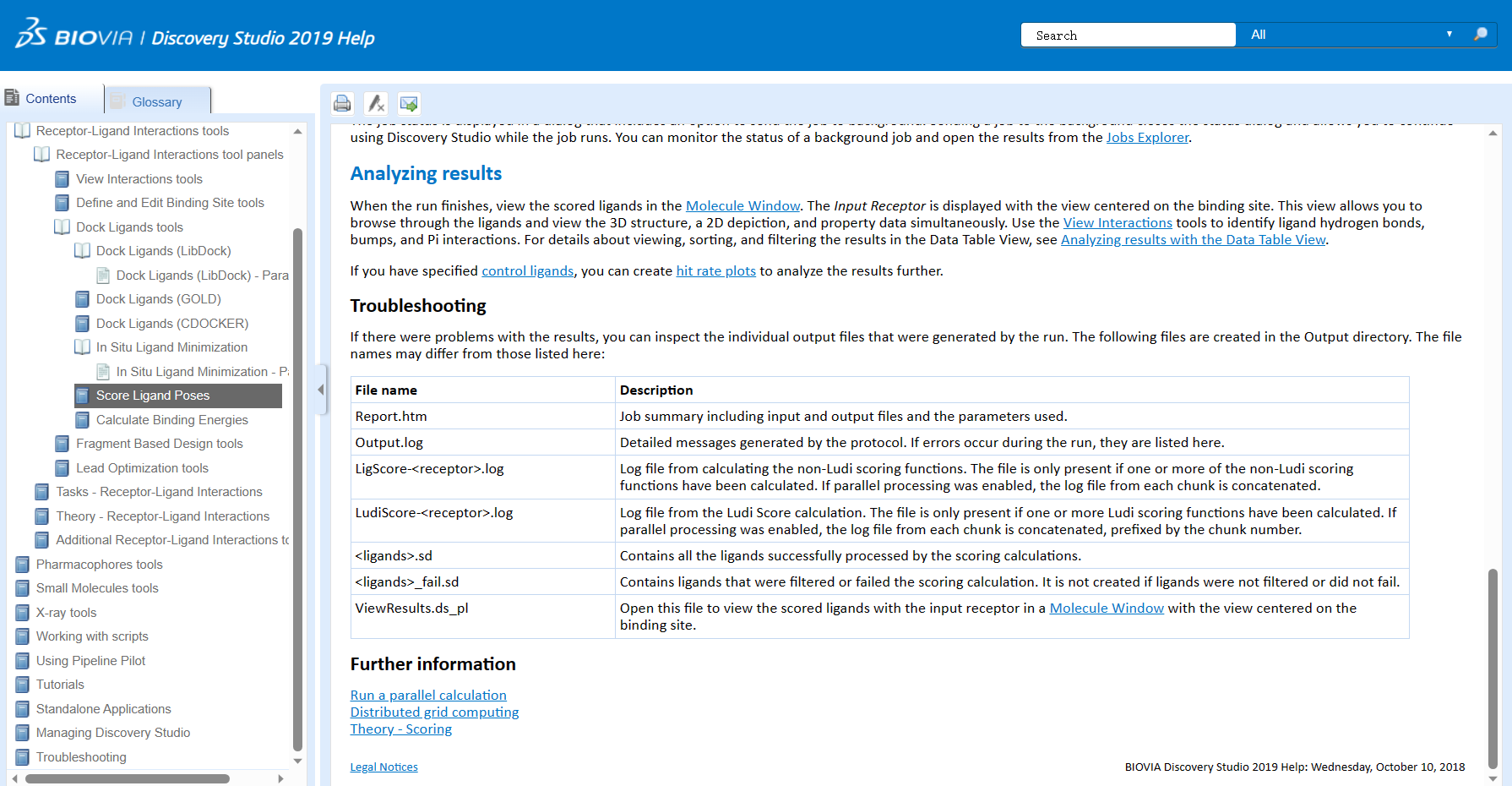
- Calculate Binding Energies: 这个功能用于计算经过对接和选择的配体构象与蛋白质之间的结合自由能或结合亲和力。它可以使用不同的方法,如MM-GBSA (Molecular Mechanics-Generalized Born Surface Area) 和 MM-PBSA (Molecular Mechanics-Poisson-Boltzmann Surface Area) 来估计结合自由能。这些方法考虑了蛋白质-配体复合物中的溶剂极化、电荷等效和分子力学等因素,从而提供结合自由能的估计值。
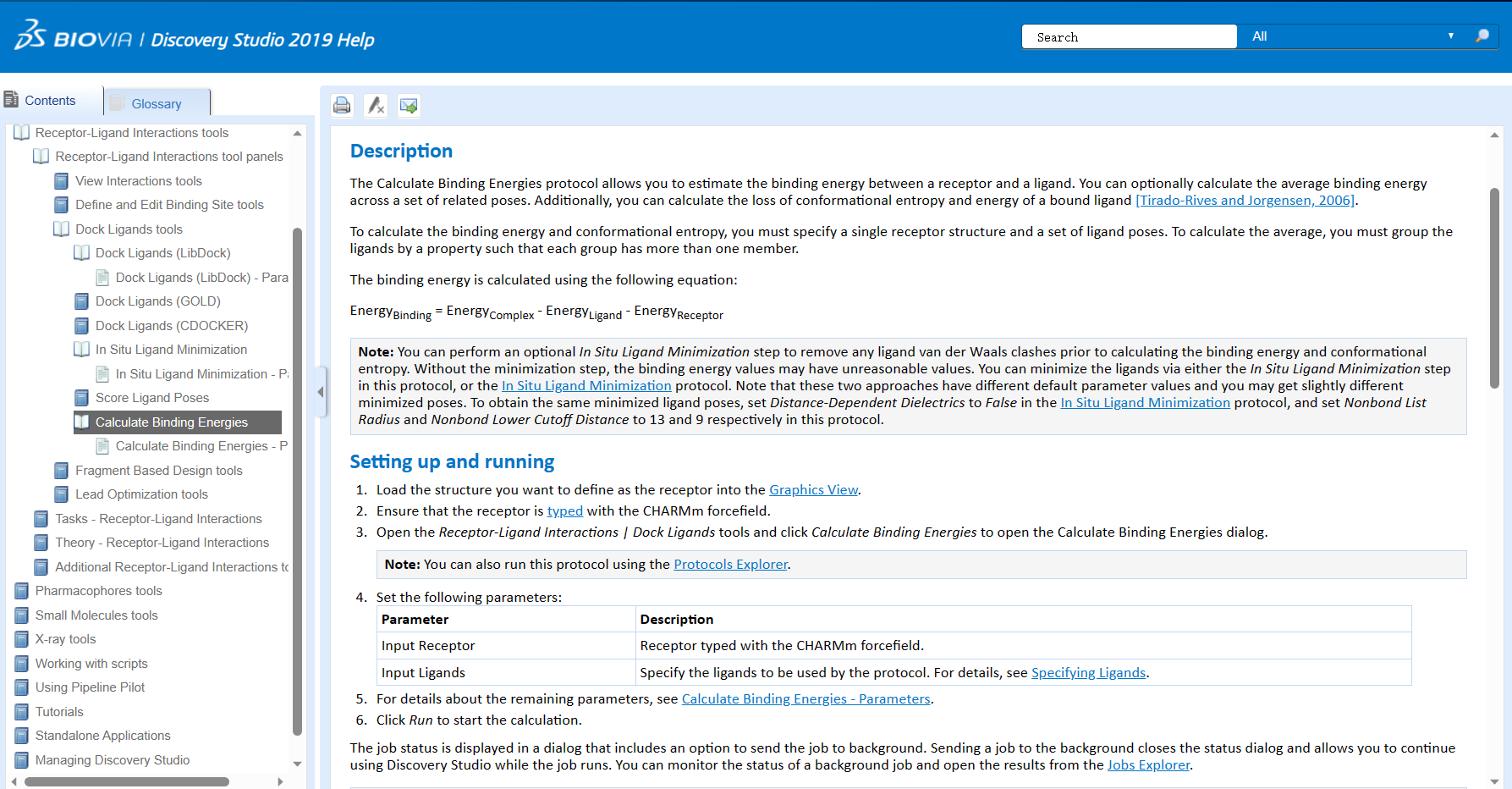
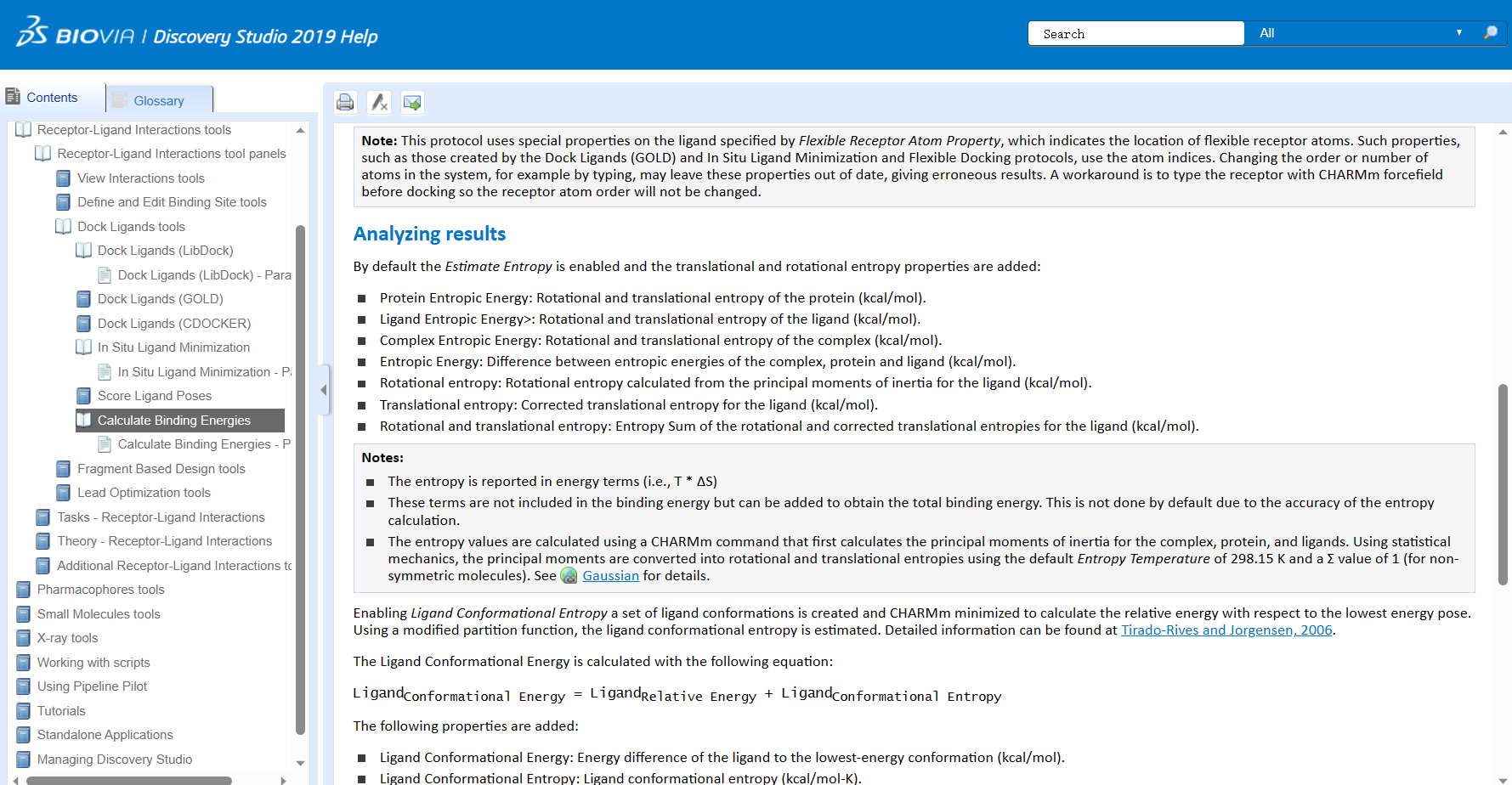
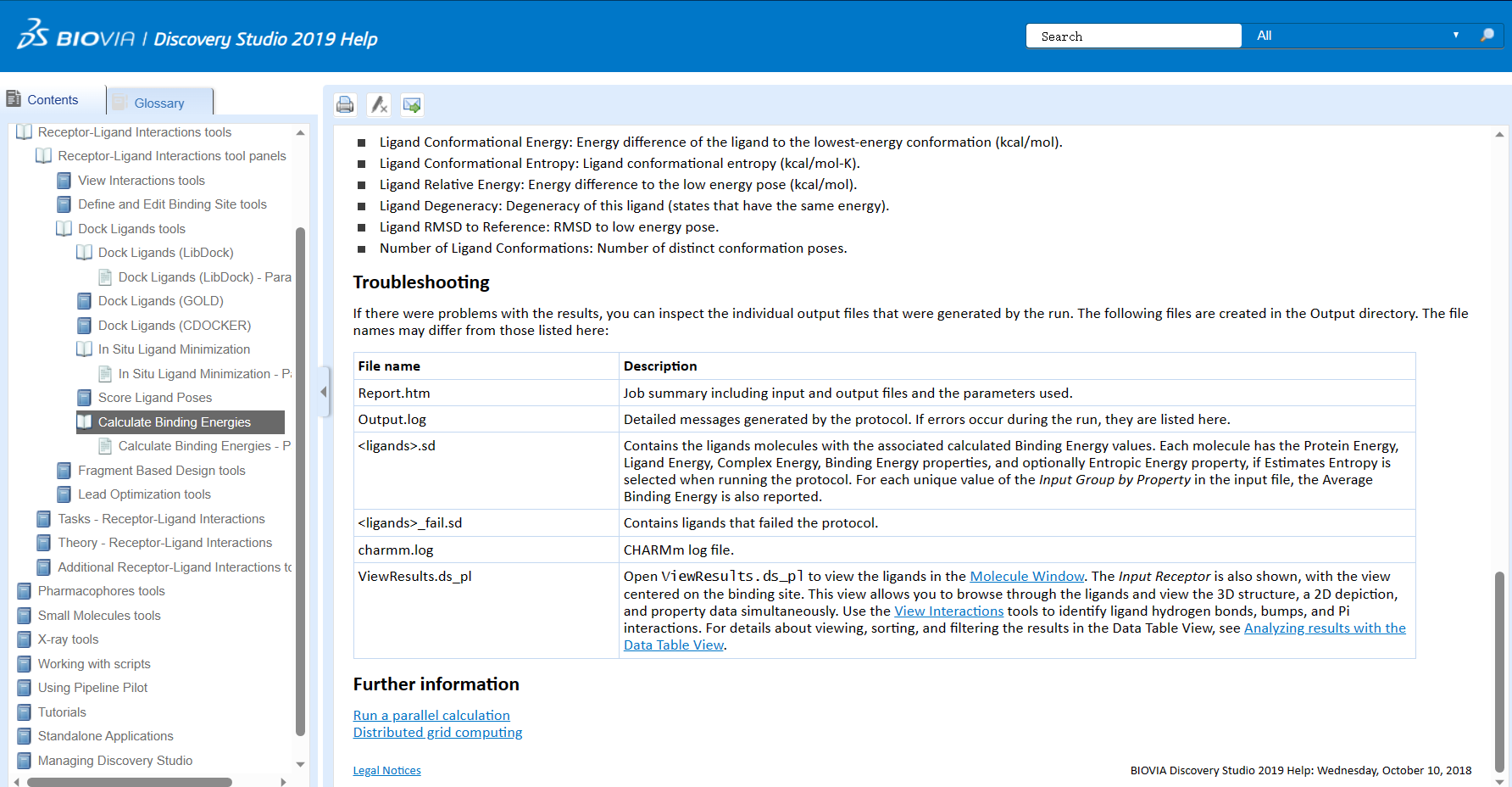
分子对接的结果如何分析以及这两个功能具体如何使用,小果将在下一期的分享中给大家带来详细的攻略,这里就先给大家留一个小悬念啦~
Define Interaction Site
通过”Define Interaction Site”功能,我们可以在蛋白质模型中选择一个或多个残基(氨基酸)定义为结合位点,这些残基通常位于蛋白质的活性位点或者与配体结合相关的区域。结合位点是蛋白质上与配体相互作用的区域,通常是药物设计和分子对接的关注点之一。
那么小伙伴们可能会问,定义结合位点有什么作用呢?
- 识别关键氨基酸残基:通过定义相互作用位点,可以确定与配体结合相关的关键氨基酸残基。
- 预测结合位点:Discovery Studio 可以利用结合位点预测算法,根据蛋白质结构和配体信息,预测可能的结合位点。这有助于我们在设计配体或者药物时能够更好地选择合适的结合位点。
- 界定结合模式:定义相互作用位点后,可以使用 Discovery Studio 提供的多种分析工具来研究蛋白质与配体之间的具体结合模式。这些工具可以帮助我们了解结合的强度、空间构型、氢键、疏水作用等信息。
- 预测互作模式:通过定义相互作用位点并结合其他的分析功能,我们可以预测蛋白质与配体之间的互作模式,包括静态结构和动态变化。这对于研究蛋白质功能以及设计新的药物具有重要意义。
通过定义结合位点,我们可以提高对药物候选物与蛋白质之间相互作用的理解,有助于更精确地进行分子对接、虚拟筛选和药物优化等工作,从而推动药物发现和设计的进展。
以下是小果搜集到的官方文档的资料,因为内容比较多,小果在这里给大家把大图展示出来:





以上就是小果给大家带来的dock ligands模块中各个功能作用的全部内容啦,在下一次的分享中小果还会给大家带来如何查看分子对接结果的详细教程,小伙伴们记得持续关注哦~如果小伙伴们平时在生信分析的操作过程中遇到困难,欢迎大家使用小果开发的生信工具平台http://www.biocloudservice.com/home.html哦。我们下次再见,拜拜~