前言
单细胞多样本整合工具大家也学过很多了吧!但是除了 Seurat、SAUCIE 和 Scanorama 之外,其他几种快速程序(deepMNN、BBKNN、Harmony、scVI、scANVI 和 DESC)都侧重于提取联合嵌入,而不返回调整后的基因表达矩阵。随着对样本水平分析的需求日益增长,缺乏调整后的表达矩阵限制了此类综合结果的利用,削弱了其有效性和普适性。因此,下一代图集级整合算法应能整合大量研究,生成共识细胞类型图和调整表达矩阵,以便进一步进行下游分析。为此, scMerge2应运而生,这是一种可扩展的高容量算法,可对图集规模的多样本多条件单细胞研究进行数据整合。下面跟着小果来学习一下吧!
scMerge2介绍
scMerge 算法可去除批次效应,并对单细胞 RNA-Seq 数据进行归一化处理。它由三个关键部分组成,包括
• 确定稳定表达基因(SEG)作为 “阴性对照”,以估计不需要的因素;
• 构建伪重复,以估计不需要的因素的影响;
• 使用快速 RUVIII 模型调整存在不必要变异的数据集。
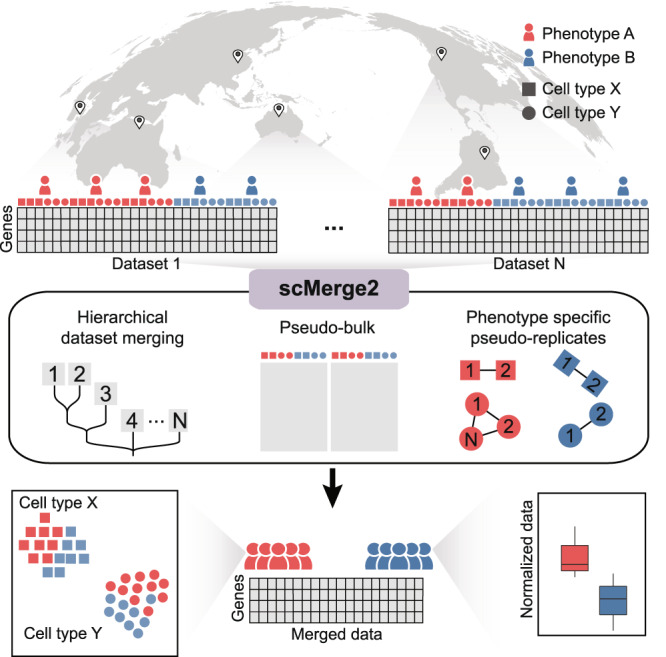
scMerge2示例
安装R包
scMerge可在bioconductor (https://bioconductor.org/packages/scMerge) 上使用。您可以使用以下方法安装它:
## 从 Bioconductor 安装 scMerge,需要 R 3.6.0 或以上版本
BiocManager::install(“scMerge”)
##您也可以尝试安装 ScMerge 的 Bioconductor 开发版
BiocManager::install(“scMerge”, version = “devel”)
加载包和数据
suppressPackageStartupMessages({
library(SingleCellExperiment)
library(scMerge)
library(scater)
})
我们在R包中提供了一个小鼠胚胎干细胞(mESC)数据示例,以及一组预先计算好的稳定表达基因(SEG)列表,用作阴性对照基因。
完整的、未经规范化的 mESC 数据可在这里找到(https://www.maths.usyd.edu.au/u/yingxinl/wwwnb/scMergeData/sce_mESC.rda)。R包附带了该数据的子样本、两个批次版本(命名为“batch2”和“batch3”,以便与完整数据保持一致)。
## 小鼠 ESC 数据子集
data(“example_sce”, package = “scMerge”)
data(“segList_ensemblGeneID”, package = “scMerge”)
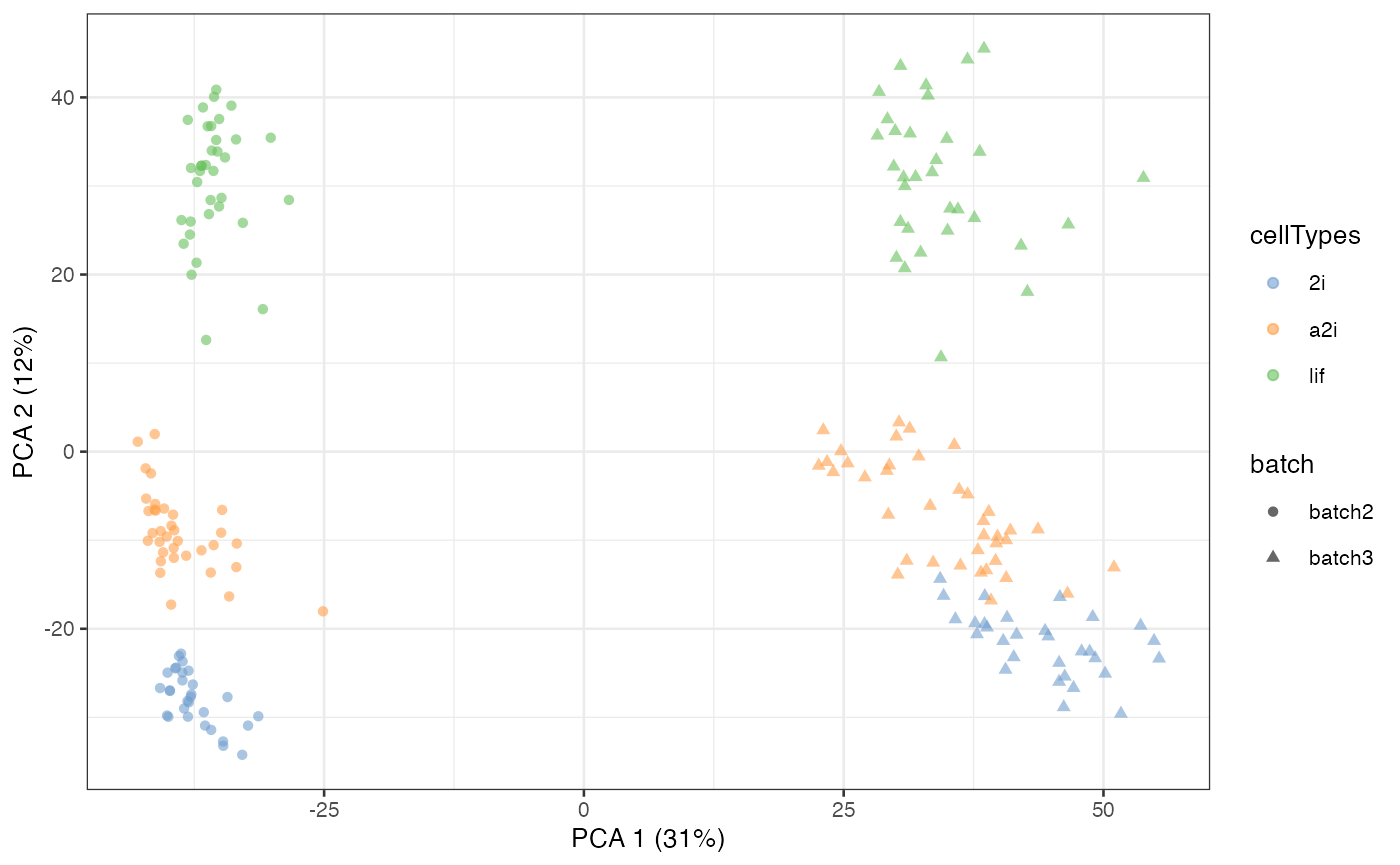
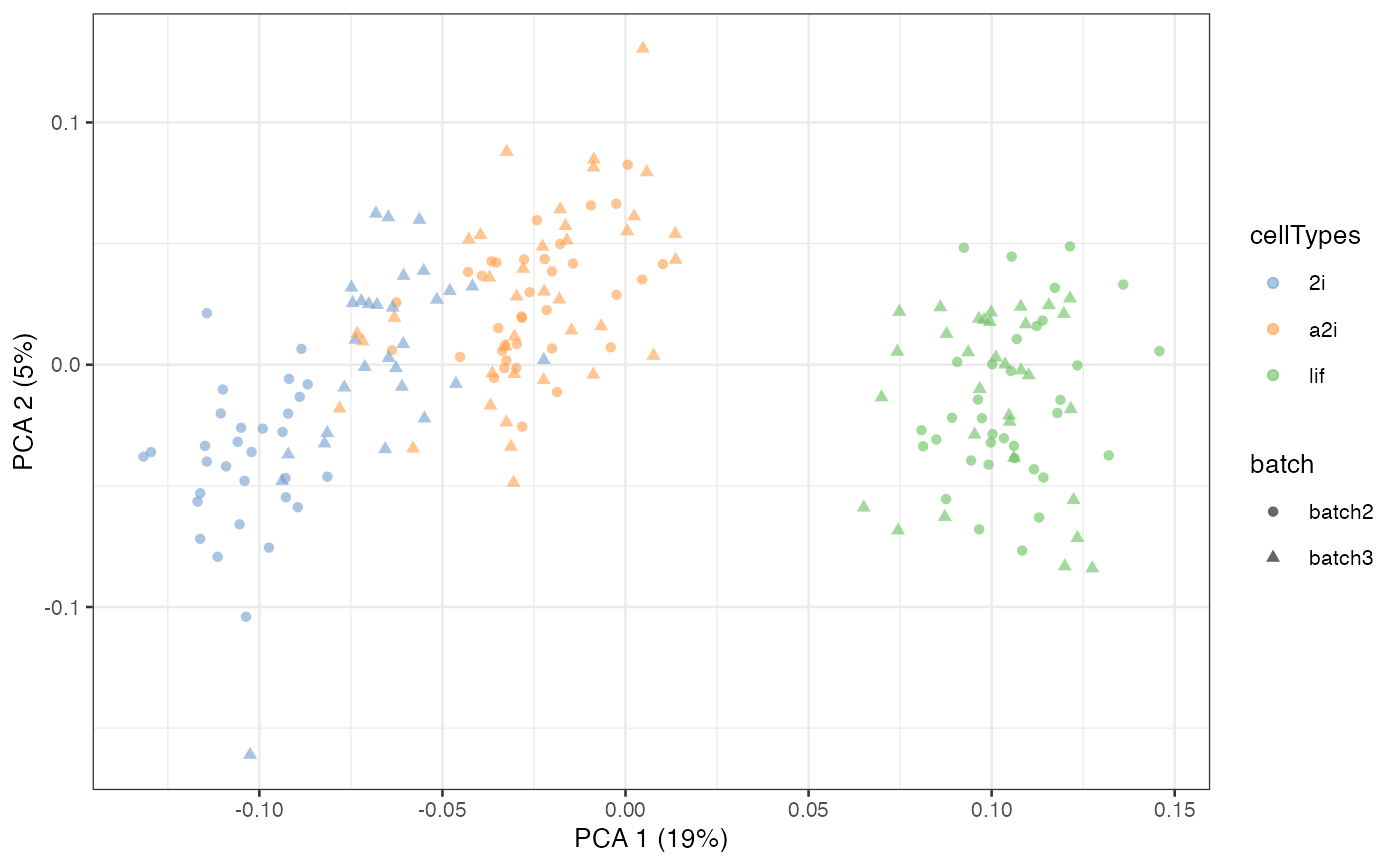
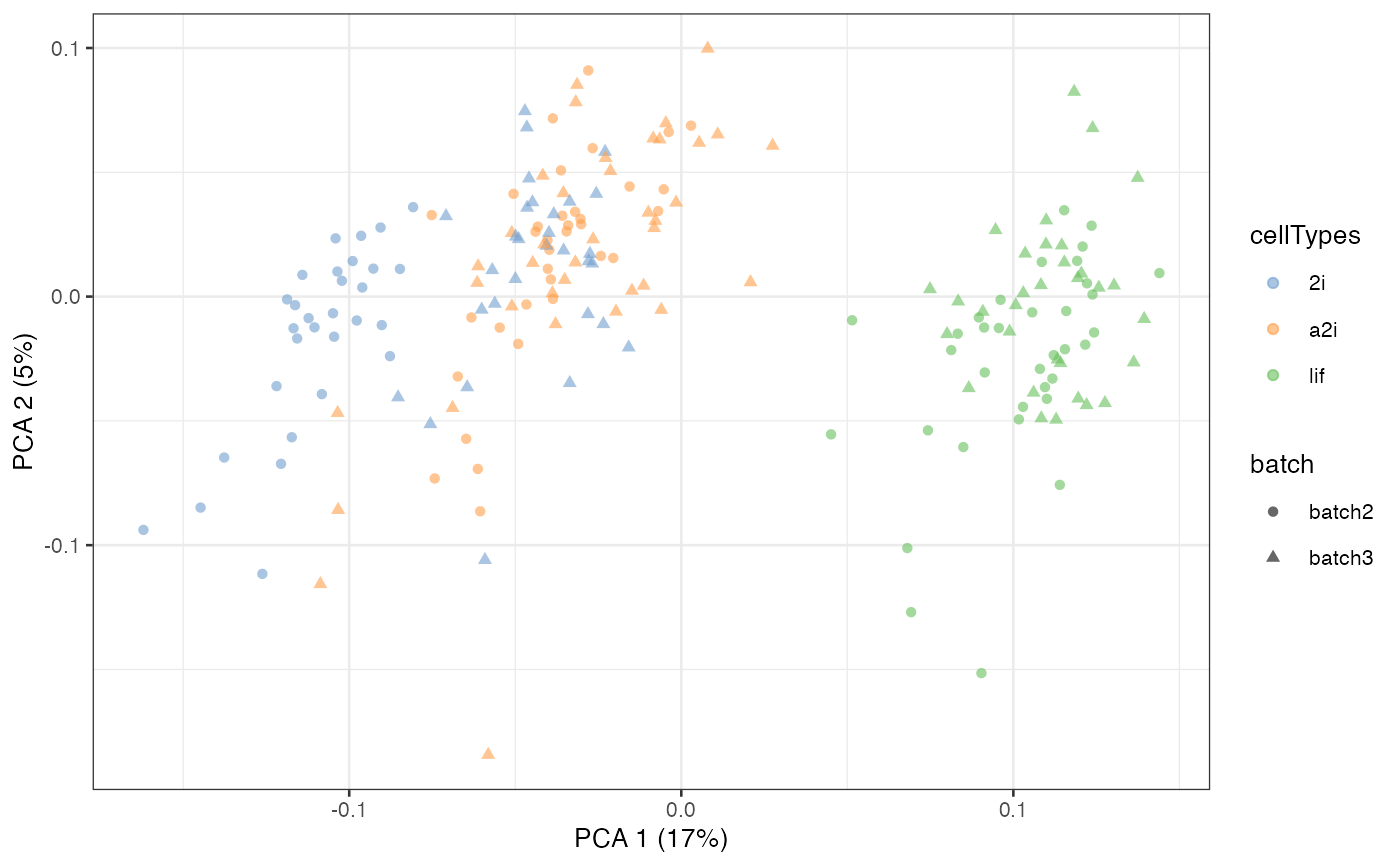
在这个 mESC 数据中,我们汇集了来自三种不同细胞类型的两个不同批次的数据。通过 PCA 图,我们可以看到,尽管细胞类型有很强的分离性,但批次效应也造成了很强的分离性。
example_sce = runPCA(example_sce, exprs_values = “logcounts”)
scater::plotPCA(example_sce,
colour_by = “cellTypes”,
shape_by = “batch”)
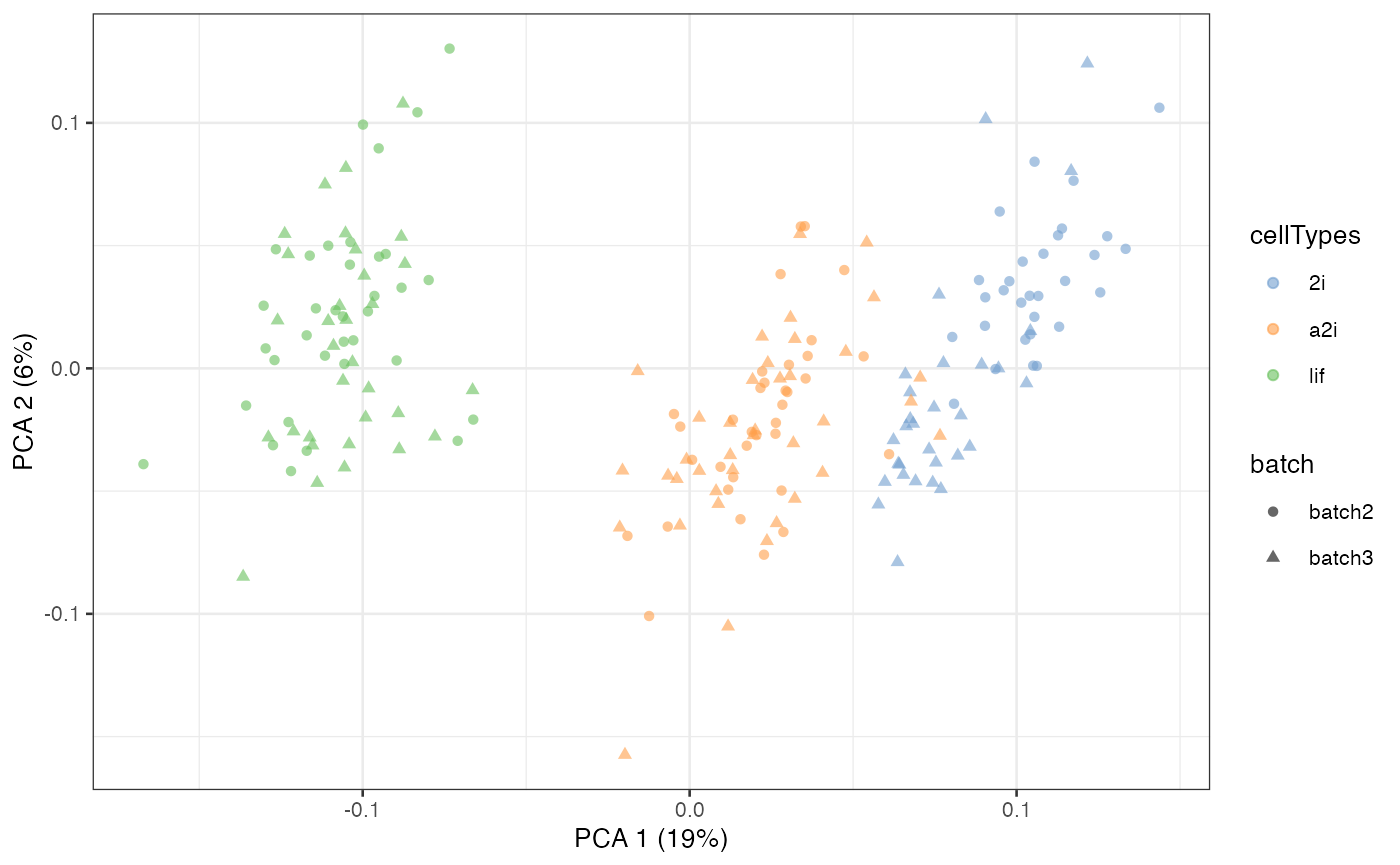
无监督 scMerge2
在无监督情况下,我们将对每个批次中的共享近邻图进行图聚类,以获得伪副本。这需要用户提供一个向量,其中包含每个批次中构建最近邻图时的邻图数量。默认情况下为10。
scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),
batch = example_sce$batch,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
verbose = FALSE)
assay(example_sce, “scMerge2”) <- scMerge2_res$newY
set.seed(2022)
example_sce <- scater::runPCA(example_sce, exprs_values = ‘scMerge2’)
scater::plotPCA(example_sce, colour_by = ‘cellTypes’, shape = ‘batch’)
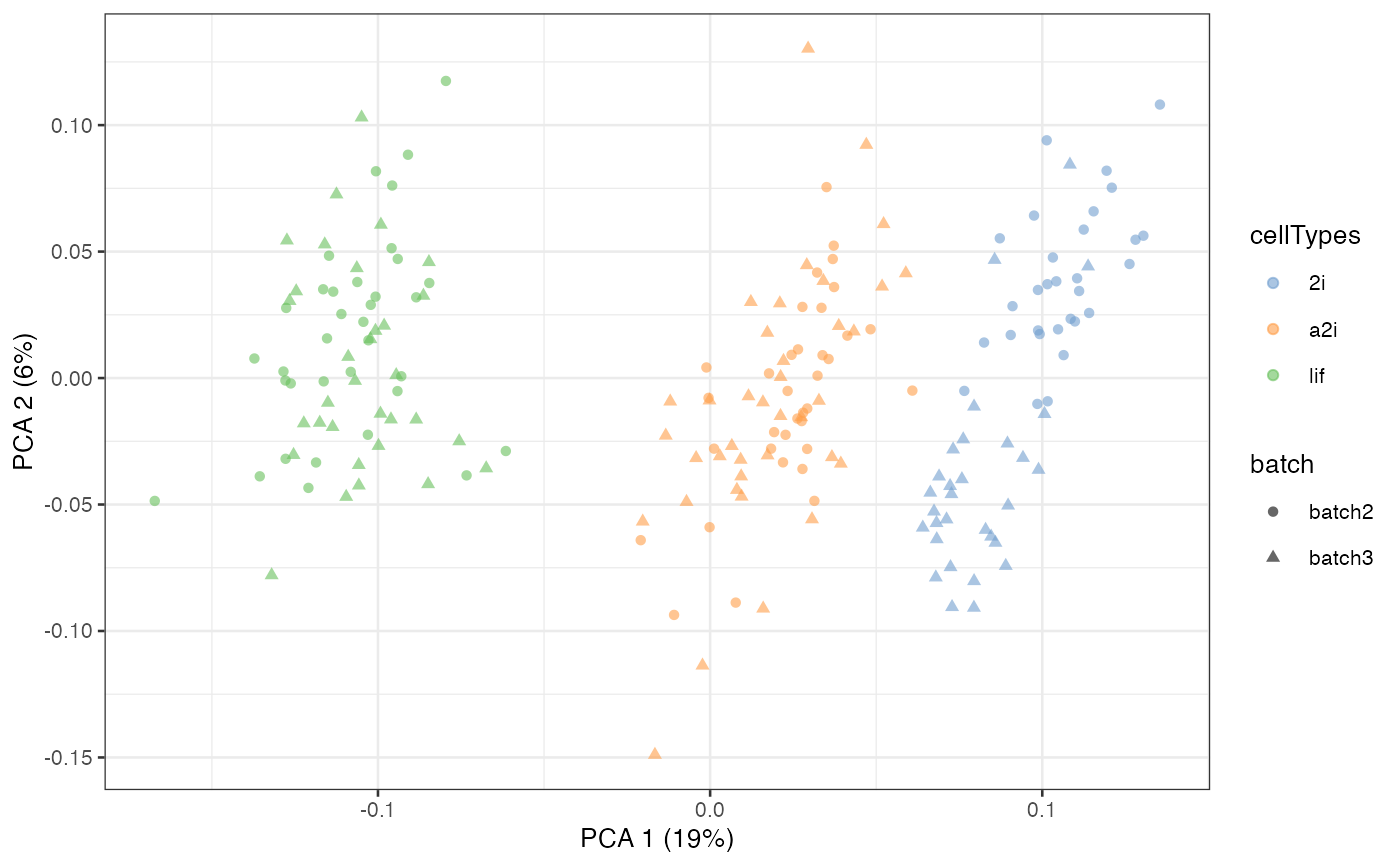
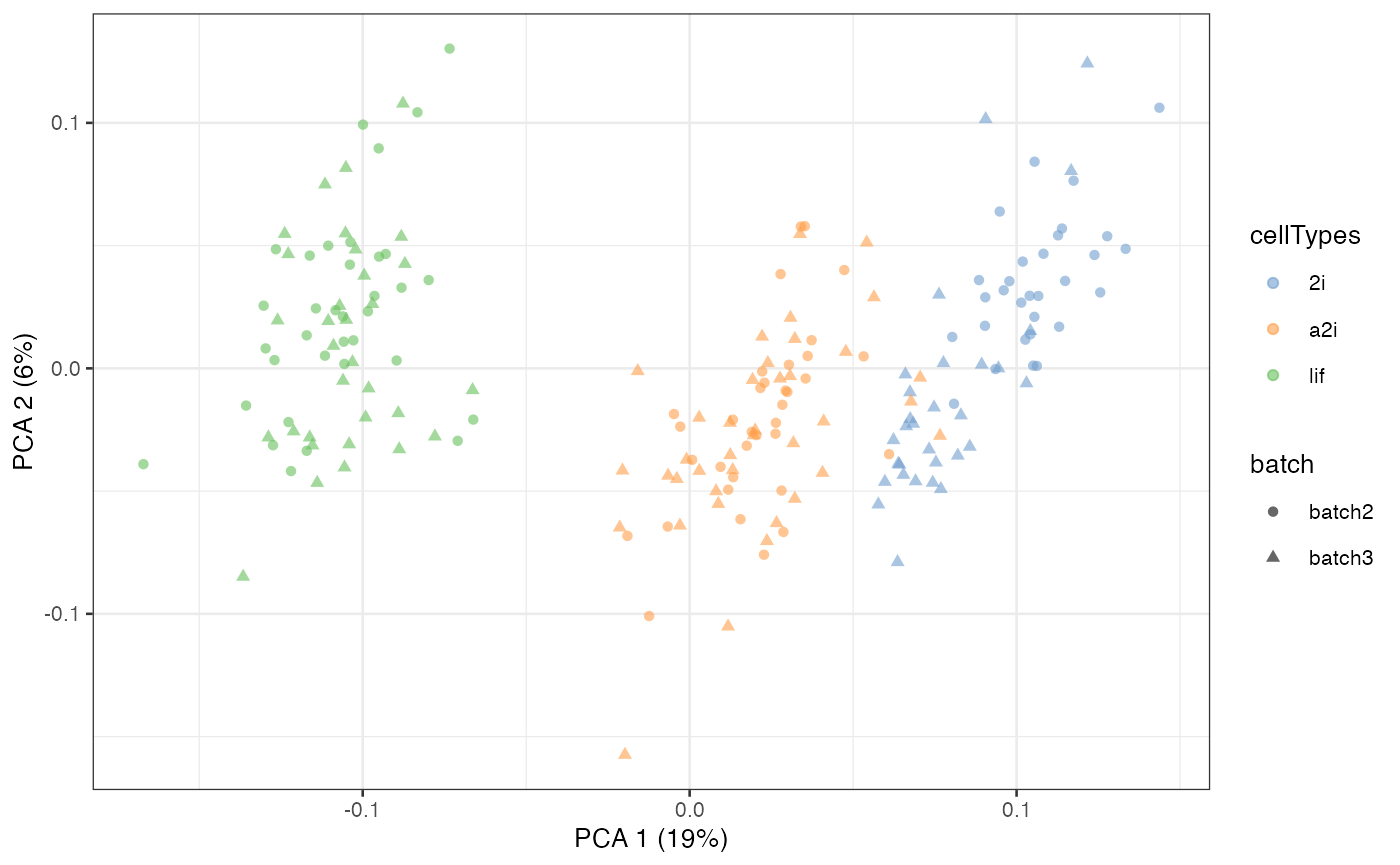
半监督scMerge2
scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),
batch = example_sce$batch,
cellTypes = example_sce$cellTypes,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
verbose = FALSE)
assay(example_sce, “scMerge2”) <- scMerge2_res$newY
example_sce = scater::runPCA(example_sce, exprs_values = ‘scMerge2’)
scater::plotPCA(example_sce, colour_by = ‘cellTypes’, shape = ‘batch’)
scMerge2 的更多详细信息
每个单元格分组内构建的伪块数量默认设置为 30。数字越大,模型估算中的伪块数据就越多,估算时间也就越长。
scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),
batch = example_sce$batch,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
k_pseudoBulk = 50,
verbose = FALSE)
assay(example_sce, “scMerge2”) <- scMerge2_res$newY
set.seed(2022)
example_sce <- scater::runPCA(example_sce, exprs_values = ‘scMerge2’)
scater::plotPCA(example_sce, colour_by = ‘cellTypes’, shape = ‘batch’)
在处理大量数据时,我们可以每次获取较小单元格子集的调整后矩阵。这可以通过在in函数中设置,函数将不返回调整后的整个矩阵,而是输出估计值。然后,为了使用估计值得到调整后的矩阵,我们首先需要对对数矩阵进行余弦归一化处理,然后计算余弦归一化矩阵的行向(基因向)平均值(这是因为默认情况下,在步骤之前会对对数归一化矩阵进行余弦归一化处理)。然后,每次我们都可以用它来调整单元格子集的矩阵。
scMerge2_res <- scMerge2(exprsMat = logcounts(example_sce),
batch = example_sce$batch,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
verbose = FALSE,
return_matrix = FALSE)
cosineNorm_mat <- batchelor::cosineNorm(logcounts(example_sce))
adjusted_means <- DelayedMatrixStats::rowMeans2(cosineNorm_mat)
newY <- list()
for (i in levels(example_sce$batch)) {
newY[[i]] <- getAdjustedMat(cosineNorm_mat[, example_sce$batch == i],
scMerge2_res$fullalpha,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
ruvK = 20,
adjusted_means = adjusted_means)
}
newY <- do.call(cbind, newY)
assay(example_sce, “scMerge2”) <- newY[, colnames(example_sce)]
set.seed(2022)
example_sce <- scater::runPCA(example_sce, exprs_values = ‘scMerge2’)
scater::plotPCA(example_sce, colour_by = ‘cellTypes’, shape = ‘batch’)
scMerge2 为需要通过函数进行多级调整的数据整合提供了分级合并策略。例如,对于有多个样本的数据集,我们可能希望先消除数据集内部的样本效应,然后再与其他数据集整合。下面,我们将说明如何建立分层合并顺序,作为scMerge2h()scMerge2h() 的输入。
为了便于说明,我首先为样本数据创建了一个假样本信息。现在,每个批次都有两个样本。
# 创建一个假样本信息
example_sce$sample <- rep(c(1:4), each = 50)
table(example_sce$sample, example_sce$batch)
要执行合并,我们需要创建一个分层索引列表scMerge2h
1.一个分层索引列表,表示要合并的单元格的索引;
2.一个批次信息列表,表示每次合并的批次信息。
场景1
我们将首先说明两级合并的情况,其中第一级是指在每一批中去除样本效应,第二级是指两批的合并。
首先,我们将构建分层索引列表。分层索引列表是一个列表,它指出了每一层中哪些单元格的索引要进行合并。列表元素的数量应与合并层级的数量相同。对于每个元素,它应包含每个合并单元的索引向量列表。
# 构建分层索引列表
h_idx_list <- list(level1 = split(seq_len(ncol(example_sce)), example_sce$batch),
level2 = list(seq_len(ncol(example_sce))))
在第 1 层,我们将执行两次合并,每一批合并一次。因此,我们有一个由两个索引向量组成的列表。每个矢量表示各批次单元格的索引。
h_idx_list$level1

在第 2 层,我们将执行一次合并,合并两批数据。因此,我们有一个索引向量列表,表示完整数据中所有单元格的索引。
h_idx_list$level2
接下来,我们需要创建批次信息列表。
# 构建批次信息列表
batch_list <- list(level1 = split(example_sce$sample, example_sce$batch),
level2 = list(example_sce$batch))
我们可以看到标有合并级别的batch_list。
batch_list$level1

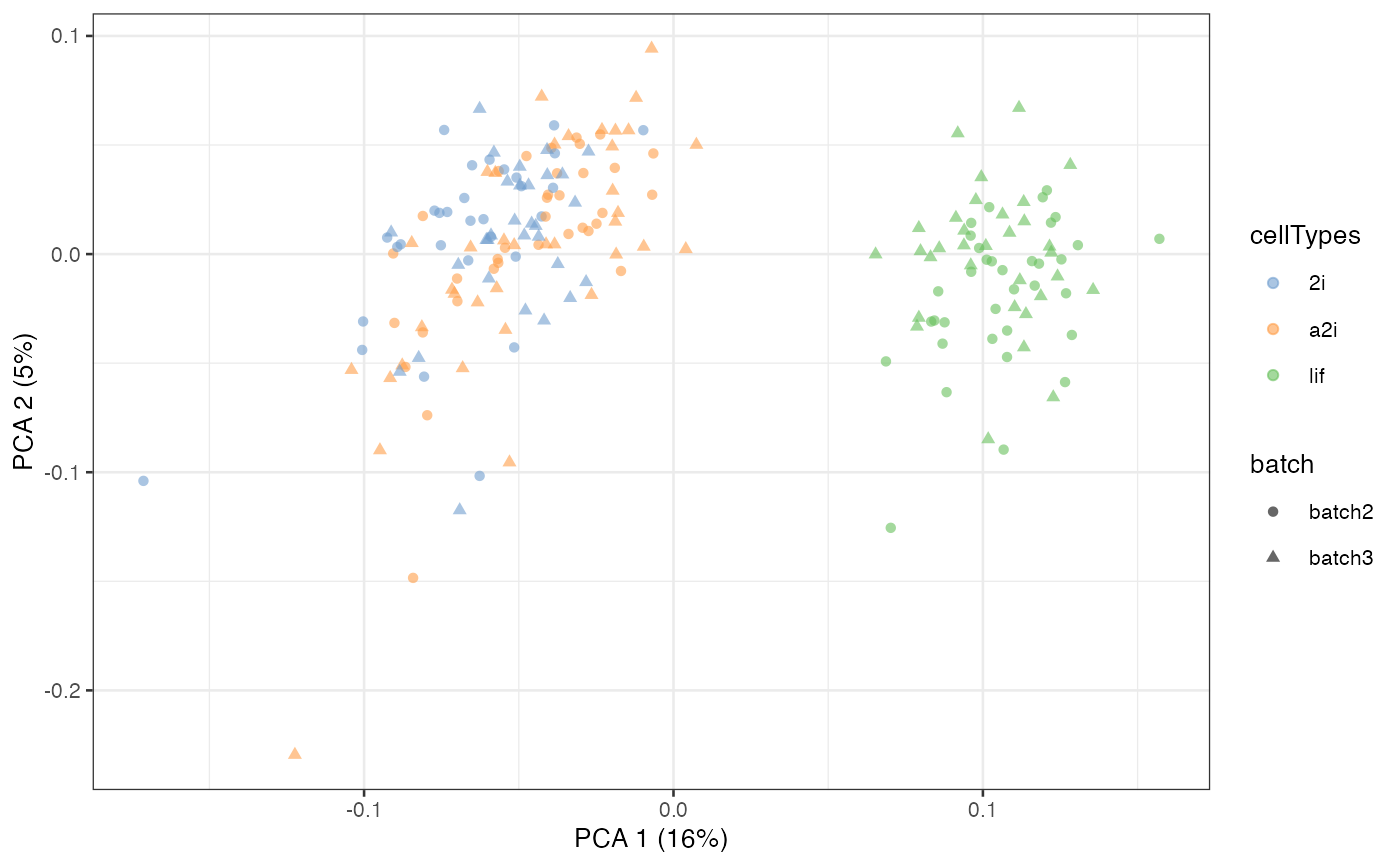
现在,我们可以输入and来分层合并数据。我们还需要输入 a ,它是每个合并层级的无用变异数(RUV 模型中为 k)的向量。我们建议在第一层输入较低的。在这里,我们将 RUV 的 k 设置为第一层为 2,第二层为 5。batch_listh_idx_listscMerge2hruvK_listruvKruvK_list = c(2, 5)
scMerge2_res <- scMerge2h(exprsMat = logcounts(example_sce),
batch_list = batch_list,
h_idx_list = h_idx_list,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
ruvK_list = c(2, 5),
verbose = FALSE)
scMerge2h 的输出是一个矩阵列表,显示每一级调整后的矩阵。
length(scMerge2_res)
lapply(scMerge2_res, dim)
在此,我们将使用上一级的调整矩阵作为最终调整矩阵。
assay(example_sce, “scMerge2”) <- scMerge2_res[[length(h_idx_list)]]
set.seed(2022)
example_sce <- scater::runPCA(example_sce, exprs_values = ‘scMerge2’)
scater::plotPCA(example_sce, colour_by = ‘cellTypes’, shape = ‘batch’)
场景2
scMerge2h 可以处理灵活的合并策略输入。例如,在上述第 1 层,我们只能合并一个批次的数据。例如,我们可以从修改批次索引列表和分层索引列表开始,删除第 1 层的第 2 批列表。
h_idx_list2 <- h_idx_list
batch_list2 <- batch_list
h_idx_list2$level1$batch2 <- NULL
batch_list2$level1$batch2 <- NULL
print(h_idx_list2)

print(batch_list2)

scMerge2_res <- scMerge2h(exprsMat = logcounts(example_sce),
batch_list = batch_list2,
h_idx_list = h_idx_list2,
ctl = segList_ensemblGeneID$mouse$mouse_scSEG,
ruvK_list = c(2, 5),
verbose = FALSE)
assay(example_sce, “scMerge2”) <- scMerge2_res[[length(h_idx_list)]]
set.seed(2022)
example_sce <- scater::runPCA(example_sce, exprs_values = ‘scMerge2’)
scater::plotPCA(example_sce, colour_by = ‘cellTypes’, shape = ‘batch’)
结语
scMerge2能够对大量单细胞数据进行综合分析。随着公共多样本多条件单细胞研究的可用性持续激增,scMerge2 证明了其整合超过 500 万个细胞以进行进一步下游分析的能力,从而实现有效的下游荟萃分析。scMerge2在所有主要细胞类型的预测准确率方面有了显着的提高。最后,scMerge2能够结合从各种单细胞技术(如CITE-seq,CyTOF和图像质谱细胞术)获得的数百万个细胞的数据。
好了,小果带着大家已经学习了80%的知识了,剩下的内容就交给小伙伴们了!最后,大家如果对单细胞数据分析还不熟悉,又想尝试一下处理自己的数据,不妨试一下小果开发的生信云平台哦,一键出图,一键导出CNS级别的Figture!!赶快去试试吧!!点击 http://www.biocloudservice.com/home.html。
参考文献
- scMerge: scMerge leverages factor analysis, stable expression, and pseudoreplication to merge multiple single-cell RNA-seq datasets. Yingxin Lin, Shila Ghazanfar, Kevin Y.X. Wang, Johann A. Gagnon-Bartsch, Kitty K. Lo, Xianbin Su, Ze-Guang Han, John T. Ormerod, Terence P. Speed, Pengyi Yang, Jean Y. H. Yang. (2019). Our manuscript published at PNAS can be found here.
- scMerge2: Atlas-scale single-cell multi-sample multi-condition data integration using scMerge2. Yingxin Lin, Yue Cao, Elijah Willie, Ellis Patrick, Jean Y.H. Yang. (2023). Our manuscript published in Nature Communications can be found here.